



Rediscovering Biology: Molecular to Global Perspectives
HIV and AIDS Expert Interview Transcript: David Weiner, Ph.D.
 Associate Professor of Pathology and Laboratory Medicine
Associate Professor of Pathology and Laboratory Medicine
Weiner is an associate professor in the Department of Pathology & Laboratory Medicine and the Department of Otorhinolaryngology of the University of Pennsylvania School of Medicine. He is also a member of the Center for AIDS Research at the University of Pennsylvania. His research can be divided into the exploration of novel methods for the generation of antiviral immune responses, and dissecting the molecular virology of HIV-1. Weiner’s lab was the first to identify an HIV gene that is associated with viral latency and silent infection. This should provide important clues about how silent infections are established in HIV-infected people, and how these silent infections later give way to AIDS.
Interview Transcript
Weiner is an associate professor in the Department of Pathology & Laboratory Medicine and the Department of Otorhinolaryngology of the University of Pennsylvania School of Medicine. He is also a member of the Center for AIDS Research at the University of Pennsylvania. His research can be divided into the exploration of novel methods for the generation of antiviral immune responses, and dissecting the molecular virology of HIV-1. Weiner’s lab was the first to identify an HIV gene that is associated with viral latency and silent infection. This should provide important clues about how silent infections are established in HIV-infected people, and how these silent infections later give way to AIDS.
How do we traditionally make vaccines?
There are two traditional types of vaccines. There are live vaccines, and perfect examples are the Sabin polio vaccine, the oral polio vaccine, or the famous smallpox vaccine. Both of these vaccines we take from a bottle and are actually a live organism. It replicates in our body. It is customized by our body because as it grows inside of us, each one of our cells is different from every other person’s, and so it is really a tailored vaccine. Our immune response grows to control it, and by learning how to control this weakened pathogen, it then learns how to fight off the true deadly pathogen. And so, those are live vaccines.
Now, because live vaccines grow in our bodies and are customized by our bodies, and because our immune system actually has to fight off the infection, they are really the best vaccines, immunologically-speaking. They frequently give life-long protection and frequently give broad protection. The body makes immune responses against all the proteins that are contained within these live antigens, and so they are very effective.
Then the other type of vaccine is the non-live vaccine. The most famous of these is the Salk polio vaccine, which is a killed polio vaccine that is injected and you make an immune response to this killed antigen. [Your body] learns how to see the real polio virus when you come in contact with it, and your immune system then makes antibodies and blocks the infection.
So what’s the problem? The problem is that killed vaccines aren’t spreading infections, and they are specific strains: whatever one is in the bottle, that’s the one everyone gets, and it may not be the same as the one in the wild, which can change. So non-live vaccines produce limited immune responses, frequently antibodies, part of the T-cell responses, but not all of them. While they’re the safest because we are really good at killing things, they are not necessarily the most effective immunologically and they don’t protect against many different types of infectious agents.
The live vaccines, which produce the best immune responses and give us our best protection, can be a risk because they are live and some individuals have weakened immune systems. For example, unintended people, who we didn’t want to vaccinate such as parents who are changing the diaper of their children who had gotten the oral polio vaccine, can get infected and that could be a problem. You could have reversion to a form that may cause some side effects in a very, very small number of people, or you could have side effects in people who are immune compromised. While they are the best, they are not the safest, and that is where vaccinology has stood historically.
The live virus stimulates a cellular immune response, is that correct?
There are really three types of immune responses that we normally are trying to induce to protect against infectious agent by vaccines. We try to produce antibodies, which are soluble proteins that are released by B-cells in our blood, and these soluble proteins bind to infectious agents and can directly inactivate them. However, once viruses and some bacteria, for example, are within cells, antibodies have trouble getting into cells to destroy them, and so there is a type of T-cell that takes care of that.
There are two actual T-cell cell responses that are important: T-helper cells and T-killer cells. T-helper cells are like the generals of the immune response. They tell the B-cells to make more antibodies and they also tell killer T-cells to find viruses and bacteria hiding within cells and destroy the cell, and so they wipe out the viral factory and eliminate the infection.
Non-live vaccines are very good at producing antibody-based responses, but because they are actually outside the cells of our bodies when they are injected, they are not very good; they are, in fact, exceptionally poor at teaching the immune system to make killer T-cells to find viruses or bacteria within cells and destroy those factories. Only live vaccines typically do that, and that is really another separation between those two vaccine approaches.
With the HIV virus, you really need a vaccine that is going to stimulate the killer T-cells, because it is already inside the cell, so the antibodies aren’t going to help. So you are trying to generate both responses?
With HIV we really, even after 18 or 20 years of studying this virus, are still not sure what type of immune response we need to control infection. We believe we need antibodies at some level because they will blunt the overall infection process when someone is exposed, but they probably won’t prevent the virus from getting within cells where it can set up shop and become viral factories that will continue to replicate. And for that, we’ll need T-cells, killer T-cells particularly.
A lot of live vaccines were considered important because they would produce both killer T-cells and perhaps antibody responses, whereas non-live vaccines would only produce the antibody responses. But the problem with the live vaccine approach for HIV is, of course, that they actually could revert, at least in the laboratory they have, to disease-causing forms. The risks are great with those, and they would need a lot more work before they could ever be used in a person.
Is that one of the major challenges and why we don’t have a vaccine for HIV? What are some other challenges?
HIV is kind of the textbook example of all the problems with making a vaccine. It has a very high replication rate, so initially when someone is infected, the virus spreads very rapidly and replicates very rapidly, and basically the immune system must contend with this really rapid replication and dissemination of virus, which is a problem.
It also infects many different cell types, but in particular it infects memory T-cells very early. Memory T-cells are called “memory” because they last a long time. HIV basically infects a pool of T-cells that lasts a very long time, and that is really a problem.
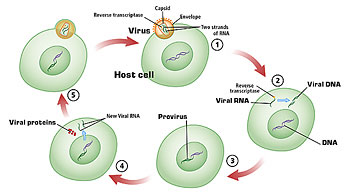
It also integrates, which means the genetic material of HIV actually is inserted into our chromosomes and becomes permanently part of the chromosomal material of that cell. Once infection happens, the only way to get rid of that infection, we believe, is to destroy that cell. Again, that would require killer T-cells.
Every time the virus goes through a single replication cycle, every time it grows, it changes a little bit, and so within a single individual there are many different variations of this virus. Then as they spread through a population, there are many different variations, and so we have to come up with a vaccine that can prevent all these different variations.
It seems like almost an insurmountable task. What is the new strategy that you have helped develop?
About 13 years ago, we started trying to develop a way to make a vaccine approach that would be as safe as a non-live killed vaccine approach. It would basically change the way these non-live vaccines work to actually also induce the really desired immune response of killer T-cells. It also has advantages of being easy to manufacture, as well as a host of other advantages.
What we turned to was to use pieces of genetic material without viral vectors so they couldn’t replicate, they could be easily manufactured in the lab, that would encode small pieces of viral gene products or bacterial gene products. The trick was whether we could develop a way to deliver them into a mouse and then eventually a person, and get the body to be fooled into taking up the small piece of genetic material and thinking that it now should make the proteins that are encoded within the genetic material.
If it did this, we reasoned that the cell that took up this genetic material would become a factory for whatever the viral protein was that we had encoded. As soon as it did that, the immune system would now see this cell as having a foreign protein, and that would look to the immune system just like a virally-infected cell, and teach it how to make the cellular immune response of killer T-cells.
But because the cell would also leak out antigen and the B-cells would see that antigen and be able to make antibodies, and because the leaked antigen would also stimulate helper T-cells, you would get basically all three components of an immune response: the antibodies, the killer T-cells, and the helper T-cells that are the essence of a live attenuated vaccine without any of the risks of spreading infection, in a non-live, non-replicating system.
Is this what is known as a DNA-based vaccine? I’ve heard the term “naked DNA.” Could you define that term?
DNA vaccines are really plasmids in most cases, in all the cases now that have gone to the clinic. We actually steal them from bacteria-they are the genetic sex factors of bacteria when they go through their mating cycle. They transfer a small circle of DNA from one bacterium to another, in order to keep up their genetic diversity, the same way all living populations need to keep up genetic diversity. That is their mechanism.
Well, these small circles are very handy. They have a sequence on them that says “grow the bacteria to large amounts.” [We] can put sequences into them to make them very specific and maintained in a way that we can regulate them and control them. And they are also very useful in that we can put other pieces of DNA inside them, and the pieces that we put in are regulatory reagents that have the instructions for making a gene product in a human cell or a mammalian cell, as well as the viral antigen.
These small circles then are highly purified from the bacteria and they have been formulated by several groups in many different ways. They are not really naked. They are actually in different solutions and formulations. Local anesthetics have been used to coat them, which help them get into cells, polyethylene glycol solutions, particles, and microbeads have been used.
But the thing that makes them all the same is they are all non-viral or nonliving, and they don’t contain protein structures that are associated with living infectious agents, and so they are just the genetic material on its own.
So you actually take one or more genes and alter the plasmid with those genes, the code for the antigenic proteins?
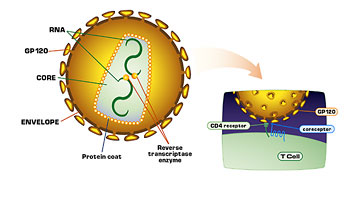
Right, we put in antigen proteins, and we also, in the case of HIV, depending on the gene, can completely destroy its function. So if it were ever to meet up with a real virus inside a person, it couldn’t help that virus in any way. In fact, if it would interact with the virus it might even prevent its replication. It might even have a negative effect on the virus. You take these pieces and you include pieces of, let’s say, the envelop, the outer portion of the virus, and that hopefully will produce antibodies and killer T-cells. Then we can also use the core antigens of the virus, the matrix, as a part of the replicative enzymes, some of the accessory genes that are part of this virus, and let the T-cells get used to seeing all of the virus.
You specifically exclude genes that would enable the pathogen to reconstruct itself and cause disease, and that is a key part of this?
That is very important. We don’t include the promoters, which are called the genetic regulatory sequences of the virus, which are at the far right-hand end and left-hand end of the genetic sequence, which are really important. The virus needs them to replicate. We also delete essential functions in something that is going to go into people such as the protease genes, which have destruction in them, and the reverse transcriptase genes. We also rearrange multiple components of the virus to make sure that it can’t reshuffle itself into some novel fashion, and we can test these laboratory assays.
What bacteria do you use for the plasmids?
The bacteria that is used is a very highly developed strain of bacteria, but where they all came from, all the bacteria that we use in most of our genetic manipulations, really are gut bacteria that originally came from people, E. coli. It is not the E. coli that we associate with disease from contaminated meat, for example. It is a laboratory adapted strain, and it is a strain that isn’t pathogenic.
How are these vaccines delivered into the body?
There are three basic ways that people have delivered DNA vaccines into the body. One is a device called the gene gun, in which the DNA is precipitated on micro-size gold beads, and actually shot into the outer layers of the skin by this device called a gun, which is really not a gun. It is just a pressure device that shoots the gold beads into the outer layers of the skin.
The other way that’s our way of actual delivery, is really to mimic the traditional old-style vaccines because we are very interested in delivery of vaccines to the developing world, and the technology has already been used across the globe is needle and syringe. Ours are formulated so they just can be injected with traditional needle and syringes.
The third way is there are topical deliveries of DNA vaccines that some groups are studying where they have formulations that can be put directly into the skin or injected directly into the skin.
So you are actually injecting the genetically altered plasmids-it is just the plasmids in a liquid form?
It is plasmids in liquid with a formulation such as local anesthetic or a microbead to get it across the outer membrane and get it taken up by cells in a more efficient way. The delivery of plasmids actually has another advantage. One of the problems with multiple boosting with vaccines, when you give a vaccine multiple times, is that you start developing an immune response to the vaccine, and then the boosting doesn’t work so well because the immune system really prevents any real seeing of the foreign antigen now. The immune system is too quick, it wipes out the antigen as it is getting into our body, and so three or four boosts, we start to not see continued boosting of immune responses, and this is a problem with some vaccines.
DNA vaccines get around this because there is no protein, and so they can be, in theory, boosted forever, because until the DNA gets into the cell and converts- uses the enzymes of the cell to convert the DNA sequence to RNA and into protein-there is nothing for the immune system to react to.
You started with mice to test these. What is happening with the tests now?
We started in mice and we went through many years of development of formulation and then we moved to nonhuman primate studies and showed that the vaccines were safe. We published in larger primates that we actually prevent HIV infection, and in a smaller primate model we’ve shown that we can impact on viral replication, lowering viral replication, and protecting CD-4 T-cells after viral challenge with a pathogenic virus.
We’ve moved on to studies in human beings with our collaborators, and these studies have taken two forms. We’ve studied the DNA vaccines; actually, the first DNA study was from our group in HIV infected people as an immune therapy, and that was conducted here at Penn. And then a subsequent study at the National Institute of Health was in the area of normal … HIV non-infected individuals as a “punitive” prophylactic approach. As of this time, we’ve gotten up to about 170 people who have received these vaccines of ours and there has been no significant adverse events; they have been very well tolerated and they’ve been immunogenic.
And now many, many groups are in the clinic with DNA vaccines besides us and our collaborators at Wyeth. We also have Merck that is in the clinic, and many other groups are in the clinic with DNA vaccines.
You were saying your group uses it as a therapeutic vaccine as opposed to a preventative vaccine, or they are being used both ways?
We have two separate studies. The first study that actually entered the clinic was a therapeutic vaccine study here, and the second study of ours that entered the clinic, which was the first prophylactic study, was in collaboration with the NIH Clinical Center. Since that time, we have maintained both programs and we are actively studying both approaches. Those programs have grown over the years as we’ve met certain hurdles and felt that we’ve improved the potency of the vaccines, which has been the issue with DNA.
When it is used as a prophylactic vaccine, how do you counsel the patient? How do you test that it is working, because you can’t tell them to go out and have unsafe sex? How do you test that it is working?
At this point in time and through trials, we will never say that we have a vaccine that works. In fact, we tell patients that we don’t have any evidence that this will be of any value at all. I think one of the real implications of this is that people who volunteer for these vaccine trials are the true heroes here. They are really being complete good Samaritans and putting up with a lot of travel to the clinics. They are getting injected with these experimental vaccines, not just DNA, but all the vaccines that are being tested, and none of them are shown to have any efficacy at this point in time, and that has to be kept in mind.
But in some ways, the vaccine trials themselves have a tremendous benefit in some unexpected ways. I would like to bring your attention to [studies] in Thailand: in some of the vaccine studies, not DNA, but the larger-scale studies of vaccines through the counseling of the recruitment effort. As people are brought in to be part of their studies, as they were counseled, the actual attack rate of the virus in those populations dropped and made those trials more and more difficult, because you needed more and more people. But it shows that just the counseling can have a major impact and is very important.
So you are having a public health effect with the education that you are providing, so that is great. How do you actually know that the vaccine is working when you inject it into the muscle cells? I am referring to those micrographs in the Scientific American article, where you made the muscle cells glow green. How do you tell that the vaccine is working?
That was a graduate student in the lab who used a very clever approach. He actually included genes that encode for a protein that will fluoresce, green fluorescent protein, and this protein taken up by cells actually is green, and then when exposed to ultraviolet light will fluoresce green, so you can see it. When you take out your biopsies and look at them under the microscope and expose it to UV light, the cells that have taken up the vaccine glow green.
What is the benefit of being able to engineer the vaccine to carry genes from different strains? Why is that so important with HIV?
With HIV and some other infectious agents like the flu, for example, the variability is really an enormous issue, the ability of the virus to change. If the virus can change in so many different ways, then the more strains we might be able to include. We might be able to prevent it; we might be able to produce a vaccine that will be protective against multiple strains.
We don’t really need a vaccine that is 100% effective to wipe out infection. One of my favorite examples of that is the Salk polio vaccine. It was really only in North America and only about 70% effective, yet it pretty much was responsible for wiping out polio from North America, and that is because so many people were protected that the virus really couldn’t spread anymore.
If we can make a collection of antigens that will prevent spread in enough people, it doesn’t have to be 100%. I believe the same thing can occur for HIV; it will be prevented from spreading and will be able to be contained, and possibly eliminated.
You are at a point now where you are taking this to the next level of effectiveness. You are using the molecules and adding it to the vaccine to make it more effective. Can you explain this?
One of the real advances of DNA vaccines is that not only can you mix different viral antigens or bacterial antigens, and not only can you combine different virus antigens if you want to make cocktail vaccines, but because it is genetic material, it allows us to bring the power of genomics, the ability to put in other genes that regulate functions exactly as part of the vaccine itself.
So instead of hunting for the right antigen that would produce the right immune response, we can actually build in the central controls of the immune system to tell the vaccine what type of immune response we want the body to make. This has really not been done before in any real fashion, and certainly not in normal healthy people in any way.
What we have done is actually tested a wide variety of immune modulated molecules, molecules that the immune system uses that we understand at different levels through basic science and think we know how they might work in people, and actually test them as part of the vaccines themselves.
For example, we include a gene called a cytokine that might expand T-helper cells as part of the vaccine and test that first in basic research studies and ask the question, “Well, does that make our protection better in some small animal challenge models? Does that do anything good for us?” If not, we’ll go on to another.
We’ve gone through this procedure now for years and years, and come up with a very small cadre of immunologically relevant molecules that now when our nonhuman primate studies produce them, they change these nonliving, non-replicating DNA vaccines into molecules able to produce an immune response, the equivalent of a live viral infection.
We think that this break-through in the primates will translate into a similar increase in immunogenicity in people, and we are very excited about that. That doesn’t mean it is going to work in people yet, of course. We get excited and then, of course, people really are where the rubber meets the road, but we are very excited that this could make a big difference and allow us to produce a very strong immune response with injections, with lower doses, and in a very controlled fashion, and produce the type of immune response we want.
So, for certain pathogens where antibodies are particularly important, we would include the instructions just for making antibodies. We don’t need to worry about T-cell responses in those individuals because in some cases you might be concerned that T-cell responses might not be good to certain pathogens.
In others such as HIV, we really want strong T-cell responses. We’ve included the instructions for expanding T-cell immunity and shown that these work in our animal models and now we are ready to test them in people. We think that this will be a magnitude jump, a 10-fold enhancement of the vaccine potency.
Once we established that they are safe in people and immunogenic, now we want to get the immunogenicity up, and increasing the immunogenicity in this way will have several effects. First of all, it will make more people respond to the vaccine on one or two injections rather than multiple injections, which is a big advantage for the patient. None of us like to take many, many injections if we can get away with one or two to do the same thing rather than five or six. That is a big advantage.
The next thing is we think it will make the immune response more consistent across individuals. And the third is because the magnitude will be so much higher, it will be a lot easier to measure and follow who we have successfully vaccinated, rather than using very more invasive tests to figure that out. So we think we can actually use very simple tests to follow people who have been vaccinated successfully and make the whole procedure a lot more friendly.
How would you do the test for successful vaccination; what would you do?
We are producing very little antibody responses, and antibody responses are very easy to follow. You take a small drop of blood and the assays, analyze a plate format, which can be automated. It is very easy to follow and quantitate those levels. Those levels may have a relationship to the T-cell response under the right circumstances.
One of the things we wanted to do was produce vaccines where we could actually see the antibody response very easily, so we could make a very rapid, very simple screen for how effective our vaccine was. These types of screens are used in the now licensed hepatitis B vaccine. Actually, the amount of antibody one produces is taken as efficacy of protective vaccination, and so these new vaccines do that in our animal models, and so we are very excited about that.
Tell me specifically what cytokines you are using.
Well the ones we’ve studied are interleukin-12, which is produced by antigen-presenting cells, cells that actually stimulate the immune response and give instructions to T-cells to particularly expand T-cell immunity.
They also affect some B-cells, however. They also do cause B-cells to respond as well, but they are really good at stimulating T-cells, and we’ve shown DNA vaccines that contain these stimulate very, very strong cytotoxic T-cell responses. They are very good at killing.
And the other cytokine genes we are very interested in is IL-15, and IL-15 actually has a lot of problems in getting expressed well. The body highly regulates it in very complicated ways for where it is expressed, and to make a DNA vaccine to use it, we had to engineer around that. [This] one is also particularly good at driving memory T-cells, so not only will the person who receives the vaccine make a strong immune response, but we believe that their immune response will last longer and that is a very important thing as well.
These engineered vaccines may have many advantages because they are sort of like designer vaccines. We hopefully are making them in a safe fashion to do what we would like them to do, and to make the whole procedure be just one injection and you would have life-long immunity, for example. That would be our dream.
I want to make sure I got this straight. You take the plasmid and you put the antigen-coding gene and the IL-12-coding gene, so you have multiple genes, then, on this plasmid?
That is correct. One of the things you can do with DNA, since to the cell DNA is DNA, it doesn’t read the DNA outside the cell; it just takes up whatever’s there. So you can actually make plasmids that have multiple genes in them, your viral antigen as well as your cytokine gene, for example, or you can mix different plasmids together to make a cocktail, and that might be particularly attractive for multiple strains of HIV.
So you would make a vaccine specific for a subtype-C, which is in Asia, and subtype-A and D, which might be in North Africa, for example, and subtype-B, which is in North America. By combining them, you wind up with a vaccine that might, in theory, produce immune responses against the vast majority of viral strains of HIV on earth.
So would you need two plasmids though? Couldn’t you put one plasmid with the different strains on it, and IL-12, and make one giant plasmid?
I love that. There is an issue of manufacturing them, and the manufacturing process where we take the bacteria that we grow in the lab in small 1.0 liter flasks and grow them; a company will grow them in several, hundred-liter flasks.
The larger the plasmids, as I said, are used by bacteria and bacteria have a big chromosome and a little circle of DNA called a plasmid. They like the little circle to be little. They start to get cranky, and so there is a size limitation. We have to think about that, to engineer it. Otherwise, they are not happy and they won’t make as many plasmid copies.
Can you define the difference between a cytokine and a chemokine?
We’ve also tested chemokines as well, so chemokines are pro-inflammatory. They are very early responders to the immune response. They are made by many different tissues and they are important in attracting lymphocytes and other cells of the immune system to certain areas. They direct traffic.
Cytokines are more in some ways like immune hormones. They actually can convert a resting T-cell to an activated T-cell or a memory T-cell, so they sort of push T-cells through developmental programs, and so that is really the difference. Whereas the chemokines really are traffic cops, they tell cells to go here or there, and wind up in certain regions, cytokines provide instructions that, you are a T-cell that we want to be now a memory T-cell, change to a memory T-cell.
To reiterate: You have tested these new augmented vaccines with the IL-12 on chimps and humans both?
Not humans. We are going to start human studies with these in the new year and we have two different studies planned, one with the vaccine trial network with our collaborators at Wyeth, and with the National Cancer Institute, and that will be normal, healthy, naive, HIV-naive individuals.
Then here at Penn, Robert McGregor will direct a study of patients who are on HAART therapy, have very low viral loads, and now are DNA vaccinated with specific cytokines in an effort to build up their T-cells, particularly their affector T-cells, and help control viral loads, and maybe even lower viral loads.
So these trials are going to help you answer some of these big questions that are left? What are some of the challenges?
Right now, DNA vaccines by themselves, even highly engineered forms without the cytokine genes, produce immune response, cellular immune responses. T-helper responses are pretty high, most people respond. But for killer T-cell responses, we are really getting less than a third of the patients to have an adequate response. What we would like to do is get that up about 85%. If we can get it to 85% or 80%, I think we would be very close to where we think we need to be for really considering testing these wide-scale.
Now, so that is where we want to go. We have to increase our number of responders in the killer T-cell type assay, or CDA T-cell assay about three-fold, three to five-fold. And if we get it up there, I think that we would really seriously consider that we’ve reached the next level.
Now there’s another trick you can do with DNA which I should point out that is through collaboration we are doing and other groups are doing as well, and that is use the DNA first as a primer, and it is mentioned in the Scientific American article, and then boost with another vector vaccine, a live vector. Those also will boost T-cell responses, and so those combinations might be very effective with a cytokine gene and then a vector boost, post the original injection.
So are you talking about a live vector virus, and HIV live virus?
No, not HIV. It’s basically one of the other approaches that other groups are using, to take a virus that has already been used for a different pathogen that is a vaccine already, and put into that vaccine HIV antigens. What will happen there is this vaccine that’s already been used in million of people now can be tested to see will it produce an immune response against HIV.
Now the problem with those approaches is because they are viral, again because they have protein antigens, and because they have many antigens that are from the virus, the other virus, not HIV. The immune system frequently has trouble seeing HIV in all that mix. Now what the DNA does in those circumstances, by giving the DNA vaccine first, is it focuses the immune response on looking for the HIV antigen, and then when you come back with the viral vector, the T-cells that see the HIV antigen now out-compete the immune response to the rest of the virus and are expanded first, and so you get very strong boosting.
Would it hurt someone with a weakened immune system at all?
You bring up exactly the right question. These are exactly questions that need to be answered and addressed, and all of those are very important issues. But that is another way that groups are considering trying to boost immune responses in an HIV vaccine setting.
What are professional antigen-presenting cells?
Well, professional antigen-presenting cells are the cells in our body that run around eating up foreign proteins. What they do after they eat them is they chew them up and they display small pieces of everything they eat on their surface in association with our matrix compatibility antigens, the transplant antigens that are a problem in transplantation.
By doing that, they basically allow T-cells to bump into them as they travel around the body or go to specific immune sites such as the spleen or the lymph nodes. T-cells then get to see these small fragments of anything they’ve eaten. If it turns out it is a foreign antigen they respond, and that is why they are called professional antigen presenting cells, because they kind of present the antigen to the T-cell. They are sort of delivery machines for the T-cell. They are frequently cells that elaborate the cytokines I spoke of that expand T-cells, so they are really the cells that make the decision on expanding T-cells, and we don’t really understand how they make the decision.
That is one of the ways that engineering DNA vaccines to contain their own cytokine genes basically makes a nonprofessional antigen presenting cell such as muscle function in some ways as an antigen presenting cell, or that it allows an antigen presenting cell that takes up the cDNA vaccine to now only make the instructions we want it to make, not make instructions that don’t stimulate an inflammatory immune response.
Isn’t one of the criticisms of an HIV vaccine, that it would stimulate killer T-cells, but that is what the virus attacks, so that might be a problem in itself?
The vaccines would require stimulating helper T-cells, which are really the cells that are attacked by HIV. Helper T-cells express a service antigen called CD4. It is a protein on their surface and it is very important in their interaction with professional antigen presenting cells.
Killer T-cells express a different antigen for their interactions called CD8, and that interacts also with professional antigen presenting cells; it is very important in their interaction. But CD4 cells are critical for expanding CD8 and B-cells, and so vaccines especially killed vaccines that expand CD4s actually could be providing cells that now could get infected by HIV.
CD8s are relatively resistant to HIV and, in fact, if we encode the cytokine instructions for expanding CD8s directly that do not require necessarily very many CD4s, the CD4s maybe will get around that problem as well. That is another reason why we are very dedicated to the engineering side. We would like to engineer a way, and we’ve actually found a different group of molecules that can do that in animal studies that are not yet ready for the clinic, that will bypass the ability of using CD4.
We’ve done and experiment with another group where we have taken a strain of mice that lacks CD4 cells completely and developed a DNA vaccine that will respond just fine, producing CD8 killer T-cells in these mice, in the complete absence of CD4. Those instructions over a few years may be very interesting, perhaps in an immunotherapy setting for HIV where really you need CD8s, and already patients may have lost some CD4s.
Your research can have some tremendous implications on the public health system, especially for developing countries: Can you explain why DNA vaccines could really be a platform of choice in developing countries where it is hard to deliver treatments and drugs?
DNA vaccines really in some ways change the way we think about making vaccines. One plasmid is really very similar in manufacturing to any other plasmid. A plasmid that contains the gene for viral protein-A is really the same in manufacturing as viral protein-B. This is clearly not the case for any other vaccine modality that we use. The hepatitis B surface antigen has completely different properties than the killed hepatitis A vaccine, and the whole manufacturing system, and every step of the way that you use in producing the facility to make those is unique, and many of the steps are unique, and, therefore, the individuals involved in manufacturing them are unique.
Some people like to say, “DNA is DNA is DNA,” and I think we are showing that by proving that we can move DNA from one organism to another. You can develop the same manufacturing for one strain of HIV or another strain of HIV, or for a prostate cancer vaccine, or for a CMV vaccine, or for a hepatitis vaccine. And because that is likely to be a lot cheaper and a lot more uniform process, that has significant implications in the developing world.
Another way that this also may have significant implications is because DNA is fairly stable, and it may be able to withstand long periods without refrigeration and that is, of course, very important in tropical environments, especially in poorer places where refrigeration may be a problem. Actually we are seeing some of that. South Africa actually at some of the AIDS meetings, has said they may want to manufacture their own DNA vaccine. Because DNA is DNA, they possibly can at least do a lot of the initial steps in doing that. They can create one that is maybe tailored specifically for their region, and grow it up, and go through purification steps in a way that is affordable. Whether they can really go all the way or not, I don’t know, but it does allow a lot more players at the table and it allows a lot more people in groups to think about very novel ways of applying this technology to public health.
In concept, it should be cheaper because it is the same platform over and over. Right now, it is not so cheap because it is relatively new. It is just like computer chips when you make the first ones, even if you say in the future they are going to be very cheap. The first ones you make are not very cheap, but once you go through the process and you start to get up to making millions of doses and everybody is very comfortable making them, I believe the price will drop dramatically.
Are they doing DNA vaccine research in some of these countries?
We are collaborating with the U.S. military on developing vaccines for Uganda, and we have another collaboration with another researcher on developing a vaccine approach for Kenya. There are other groups such as the VRC that is looking at South Africa as a place to develop DNA vaccines, and I know Merck is interested. Many groups are thinking about developing DNA vaccines for Africa, for South America, for China, and for India. So DNA is sort of becoming an infectious approach to making vaccines in many of these really important areas.
Also with malaria, there is a lot of work now in DNA vaccines through the Navy and other groups.
How far away are we from actually having a vaccine?
I believe in the next year we will learn about the envelope subunit approach of VaxGen, which those Phase 3 trials, we should start getting those data from those trials. Now that is a subunit approach, just the envelope protein itself designed to produce the antibodies, and maybe some T-helper cell responses.
Many people have felt that it is unlikely to be very effective, but we are very interested in seeing how those vaccine approaches come out, because it likely doesn’t induce very broad immune responses. But everyone is hopeful we will learn something very important from that. We’ve certainly learned, because of their pioneering efforts, how to run Phase 3 trials and get the data.
Probably following that, there will be vaccine studies of pox viruses with Aventis Pasteur, and those should follow in the next few years after that. A couple of years behind them will probably be the DNA vaccines, so DNA vaccines will probably start getting data back maybe from larger trials in five to seven years. If the earlier parts of those trials start producing very strong immune responses, I think you’ll see a large number of candidates that we are starting to feel a little more comfortable with, get to trial.
But it is really important to realize that just because we feel comfortable with them doesn’t mean we are right, and I can give you two examples of that. One is the original development of a herpes vaccine, which was done by Chiron about a decade ago. Chiron’s vaccine was a subunit approach for herpes and when Science polled scientists in the field and asked how effective this vaccine would be when the Phase 3 trials were running, overwhelmingly individuals said it was likely to be highly effective. Well, when the trials were completed it didn’t show any efficacy in those early trials.
Conversely, the original rotavirus vaccine that ultimately had some other issues associated with it, because it didn’t prevent infection completely in animal models was thought overwhelmingly not to be a very effective vaccine. Well, ultimately it was 80-90% effective in preventing death and disease.
So it is really important that we run trials, because just because we think something–and even if we are in agreement–it doesn’t mean we are going to be right. We can only learn this by going through the process.
You were saying that you are not necessarily trying to prevent this disease either; it is a therapeutic approach, right?
Right, the “floor and the ceiling” is we don’t need to be 100% effective. I believe even if we are 50% effective, we might have a significant impact in many ways. Now that will present a lot of issues for public health designs in that whole system.
There is another way that we may be very effective and cause another huge problem, and that is if the vaccines don’t prevent infection, but prevent death and disease. Then we would have a whole population of people who are infected, but won’t progress. And you might imagine that it is really staggering just to think about the challenges of managing that situation. Certainly it will be a benefit, but those benefits carry with them a whole bunch of issues that we then have to solve.
Because vaccines are down the road, how important is it to continue really strong public health policies and prevention information and education, and get that information out to people?
I think the public health approach is perhaps one of our most important weapons in this campaign against AIDS. Just the example of the vaccine trial organization dropping attack rates eight-fold or ten-fold shows you how potent they are-that’s basically dropping the attack rate 90%. If we had a vaccine that dropped the attack rate 90%, we’d be very excited. So public health can have an incredible impact. I think that should be our front line defense against HIV. There’s no issue there that it should be something we rally behind. Since we really can’t predict when we will have a vaccine-or once we have a vaccine, how effective it will be-I can’t see in the foreseeable future that we can relax at all our vigilance in the public health efforts.
There are so many advantages, it sounds like, to DNA vaccines. Could just summarize the advantages to a DNA vaccine?
The advantages are that they are simple to manipulate; they bring the power of genomics to vaccine approaches where you can build a vaccine in a very rapid and cheap fashion-the way you might want it to be-and it allows all the creativity of genomics. They’re easily scalable in manufacturing; they could be mixed as combinations. My daughter Rebecca several years ago, when she was smaller, drew a cartoon of a little girl’s arm and a vaccine-I didn’t put her up to this. She was visiting and she was drawing with crayons in my office and the vaccine was multiple colors-purple, red, green and blue, I think. I wasn’t sure what she was doing, and Jean Boyer asked Rebecca, “What are you doing there?” She says, “I’m making the vaccine that you’re making.” We asked her what that was vaccine about, and she says, well, the colors are all the good things you need, and none of the bad things that would hurt you. I think that that’s really what a DNA vaccine could really be like. We can take out the things we don’t want and put in only what we need and make them function very specifically, and in a relatively, hopefully cheap and easily reproducible fashion.
What are the really big questions-what are the open-ended questions you’re really working on now?
Some of the unanswered questions are making these vaccines very consistent from person to person. Knowing that the T-cell response is related to the antibody response magnitude so that we can use very simple Elisa-based tests and formats for it if we ever get to Phase 3 studies. Because doing Phase 3 studies on T-cell biostates is really a lot more difficult, and certainly a lot more difficult for developing world. Elisa-based assays would be much simpler. Some of the other problems are in the logistics of vaccine development and implementation. For example, in a trial site we looked at in Uganda, the roads are not permanent. They move. And so when the vaccine’s actually administered by going out to those sites, visiting with the people and taking samples and follow-up is difficult-and so the military there has come up with using the Global Positioning System to actually find where each person is, rather than a road on the map. There are all these wonderful, clever people trying to figure out ways to make these next vaccine approaches and studies and designs really implementable. And those are really challenges.
In science, I think coming up with and showing that the approaches that we’ve made induce long-term memory T-cells is a challenge. Then really coming up with an approach in nonhuman primates that completely prevents infection, which we don’t have yet.
I know there are people out there who probably say that vaccines just aren’t going to work for HIV: that you’re never going to find something that’s going to really work. What is your response to that?
I think that vaccines have taken a lot of hits across the board recently. But I think that if we look at the data: Smallpox wiped out a billion people in the last 100 years it was on this earth. That’s much more people than were killed in all of WWI, all of WWII, the Vietnam war-any way you want to stack it up. Vaccines are our number one public health success other than plumbing, really. There’s nothing that comes close. Smallpox was eliminated-except now, for the threat of bio-terrorism-from the earth. It wiped out half of Europe twice. Polio, which ravaged North America and Western Europe and many parts of the globe is near extermination-not there yet. We don’t think about vaccines in this country so much. If you just think about hospitalization visits, they have made such an enormous impact in our lives.
Now, we can’t predict the success of DNA as a vaccine approach. But because of the advances in the science, and what we see in the immune responses, maybe HIV-because HIV is tough-will take time. But I am an optimist. I believe we will get HIV to succumb. But I also believe that, along the way, we’re going to learn how to use DNA, because as we have to make it better and better for HIV, it’ll become very useful for many, many other pathogens, and also cancer approaches.
Monoclonal antibodies took about 20 years from the time they first came on the scene to actually deliver us really wonderful, useful products- such as for treating autoimmune disease. In the middle, people got very tired of them and started badmouthing them. When they came back and were successes, it was almost as if we had rediscovered them, like a lost teddy bear or something. I’m more than hopeful that DNA vaccines will be the same way. It takes a little while to get there. Science takes time, and it takes so many people. Not just scientists. It takes all these people, [including] the media. Everybody participates in bringing these things to fruition. The public. And so it takes time. But I believe we will get there.