





Rediscovering Biology: Molecular to Global Perspectives
Proteins and Proteomics Expert Interview Transcripts: Stanley Fields, Ph.D.
 Director, Center for Gene Research and Biotechnology
Director, Center for Gene Research and Biotechnology
Director of the Oregon State University’s Center for Gene Research and Biotechnology. Carrington’s lab conducts research on how viruses and host plants interact, using the model organism Arabidopsis. He uses genetic, genomic, and proteomic strategies to understand RNA silencing pathways, virus recognition events, and cellular targets for various RNA silencing suppressors. Carrington’s research in RNA silencing has been included in the 2002 scientific “Breakthrough of the Year” in the journal Science. The magazine cited a body of work being done by several research groups across the nation on small RNA molecules, calling them “electrifying discoveries, which are prompting biologists to overhaul their vision of the cell and its evolution.”
Interview Transcript
Fields is a professor of genome sciences and medicine, and adjunct professor of microbiology at the University of Washington. He analyzes the function of proteins from the yeast Saccharomyces cerevisiae on a genome-wide basis and uses this yeast to develop assays that can be applied to proteins from any organism. In 1989 Fields and his colleagues developed a methodology for finding protein interactions, called the two-hybrid system. Since that time, other methods have been developed and used with success, but Fields’ system has become the dominant tool among researchers throughout the world.
What is the difference between the genome and proteome?
Well, people often [say that] the genome is the genetic blueprint, and so if you think of this for a car-the genome being the structural diagram for the car-the proteome would be the engine, the chassis, the carpets, the lights, the steering wheel, the gearshift, and so forth. The proteome is all of the proteins, so it’s all of the functions that take place within the cell.
Genomic [research] has a big advantage [over proteomics] in that DNA from any species in any organism behaves much the same. All DNA is pretty much alike. Every protein behaves differently, so when you think about analyzing thousands of proteins the methodologies that you have to use are a lot more complicated.
The information you get in proteomics is of many, many different types so you can ask the question: Where do proteins move around in the cell? You can ask the question of what proteins other proteins are interacting with. You can ask questions about what the structures of proteins are — many, many different assays.
In genomics, you’re generally confined to one-dimensional DNA information and you can ask which genes turn on or turn off in a given cell, but you’re not able to look at all of the very different properties that proteins are capable of in a cell.
Q: How has the study of proteins changed?
A lot of which changed in the last few years is the ability to do an assay. Instead of assaying just one protein, [you can] do it on thousands of proteins at once and that requires big changes in automation, in robotics, in the ability to express more than one protein.
In trying to understand how the cell works, you want to understand lots of different processes, and each of those processes has many proteins involved and so when you could only study a few proteins you had a very narrow field of view-now you can look at the whole cell at once.
How else has the research changed?
Well, traditionally the field of biology was thought of as hypothesis-driven. You had some idea of how a particular process worked and you asked a question that would be designed to answer that specific question.
Today, many of the experiments are done in a hypothesis-free background-you’re asking the question of all the genes in the cell-which ones are expressed in a certain cell or in a certain cancer or under certain environmental conditions. Similarly, with proteins you would ask the question: Can we look at all the interactions of proteins or all of their modifications or localizations rather than homing in on just one protein?
What can knowing the proteome of a cell tell you?
It can tell you dynamic properties of the cell. It can tell you that certain processes are affecting other processes, when cells make the decisions to divide they have to do things with their chromosomes, there are all kinds of repair processes going on. There are lots of components in the cell that have to move around and the study of all of those proteins at once has a big advantage over just focusing on just one or two proteins.
What are some of the difficulties encountered in proteomics research?
What makes proteomics challenging are two different facets. One is that proteins have very unusual and different behaviors, and so once you’ve learned how to sort out conditions for one protein, it’s difficult to translate that to a second and a third and a thousandth and a ten-thousandth protein, unlike with DNA.
And secondly, proteins go through many different modifications. They go through cleavages. They have different half-lives in a cell. They go to different compartments so there are lots and lots of different variables you’re trying to deal with when you’re studying proteins.
Has the “central dogma” changed?
Well, it used to be thought that a gene would encode a single protein, and now we know that a gene might encode ten or even more different variants of that protein. These variants can differ in where they start, where they end, which amino acid residues are included, how the protein is modified by other chemical groups, and how the protein gets cleaved, so the proteome could be tenfold or more complicated than the genome is.
The most common way a gene can make ten proteins is through alternative splicing. A gene encodes an mRNA that goes on to be translated to make a protein and a gene can make many different mRNAs, which vary by splicing alternations-that is includes different pieces of the RNA sequence in the final mRNA-and each one of those messages make a different protein.
Why is the field of proteomics exciting?
It’s exciting because we now know enough about the basic cellular processes that what we’re trying to understand are more complicated features in many cases of regulation of dynamics of how proteins are changing in the cell, how small variations in proteins have big effects on disease process, for example, and proteomics allows you to get a handle on lots of different proteins at once.
What are proteomics researchers studying most of the time?
For many studies that occur in proteomics, you’re usually only studying one representative of each of those protein sets of protein families that come from a gene-we’re not at the level now that we can look at all of these thousands of variants. Yeast has about 6,000 genes and so we’re studying them as if there were 6,000 proteins. We’re just ignoring the fact that many of those proteins are just one of a whole set of different proteins from that gene.
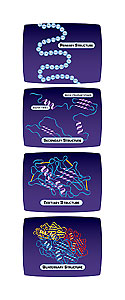
Why is knowing the 3D structure of a protein so important?
The three-dimensional structure of a protein at the detailed level is important for several reasons. One is it can tell you a lot about how a protein works-in biology, function always follows form, and the structure of a protein dictates what that protein is capable of doing. If you really want to understand [a] process like DNA replication or cell division in particularly to understand it well enough that you can understand why a disease like cancer is a change in that phenomenon. You really have to be able to look at those proteins at the level of structural detail. You have to be able to see which protein fits into another and which proteins bind to small molecules in the cell and you can only do that through the structure.
Now secondly when you’re designing drugs you want to be able to slot small molecules into protein structures and so again to be able to do that you have to have very precise knowledge of how that protein folds up in space.
If you know the amino acid sequence, does this give you the 3D structure o of the protein?
If you know the primary sequence of a protein at the level of understanding we have now, you cannot fold up that protein into a three-dimensional shape. The reason is that there are billions and billions of permutations as to how that folding can occur and we don’t understand it well enough to be able to say which is the exact one that occurs. Computationally, it’s an extremely difficult problem.
There are [now] people working on that and they’ve made a lot of headway so if a protein in its primary sequence looks like other proteins that have been solved, then it’s possible to make a model of that protein that’s pretty good.
There are other people who have come up with algorithms where they can take a primary sequence and predict a three-dimensional structure. Some of those structures are roughly right-these computational methods are getting better and better. But none of those methods will approach solving a structure at the three-dimensional level that would allow you, for example, to design a drug to fit into that protein.
Can you explain protein domains?
Proteins are modular in their structure so that when the three-dimensional structure is looked at, there are separate folded portions of the protein and often each of those portions or domains carries out a different function.
So, for example, if Protein A is going to bind to Protein B, it often does so by 50 or 100 amino acid residues folding up into a structure in A that recognizes a complementary structure in B. But Protein A might be thousands of residues long and it’s just a set of 50 or a 100 that actually carries out the A-to-B protein-protein interaction.
Then another part of A may be involved in binding to Protein C and so another part of A folds up into a different structure and that structure is complementary to C. And a third part might fold up and be complementary to D. And a part of the protein might fold up to recognize ATP or recognize some other small molecule and each of those separately folded entities constitutes a domain.
What are some of the new tools that have been developed to help the field of proteomics move forward?
The technology has changed to enable proteomics in many different ways. One is the rise of genomic information and so by having all these genome sequences for many, many different organisms including the human, you’re able to predict a parts list of proteins and so that’s changed in the last few years dramatically.
Also, to be able to interpret that genome sequence and predict proteins, computational methods have changed drastically and the amount of computing power that’s now needed in biology has changed.
[Another change is that] robotics and automation have come into play so we often are dealing with proteins in micro format-in plates in which we have 96 or 384 different wells, each well containing a different protein that’s handled by robots to move from one location on a robot to another.
Another way that the technology has changed is that instrumentation has come along. Mass spectrometry is the most notable of these that can detect really minute amounts of proteins, so if we take a mixture of proteins that might be [in] a blood sample, we can identify what those proteins are with a sensitivity that we haven’t seen before because of instrumentation.
Additionally, other methods have come along-genetic and biochemical methods-that allow us to look at protein activities in ways that we couldn’t do until just the last few years.
You mentioned a mass spectrometer as an important proteomics tool: can you explain why its role is so important?
What the mass spectrometer does is tell you the mass of a protein or more typically the mass of a peptide, smaller fragments of a protein. It tells you the mass with such precision that if you know all of the predicted proteins of an organism, you can theoretically calculate what the masses of all the different peptides would be. Then when you take a [sample] of proteins and chop them up into these peptides and put it into the mass spectrometer, you’ll get back these sets of masses, which are compared to all of the possible masses of peptides from that particular proteome. You know immediately when you get a match-or the computer tells you you’ve got a match-you now know what peptides and hence what proteins are present in that sample.
So if you’re studying Protein A, and you purify A as part of a complex with Proteins B, C, D, E, F-you want to know what all those proteins are because you want to understand the function of the complex. So you can chop up A, B, C, D, E, F into these different peptides, feed it into the mass spectrometer, and you’ll get back a series of peptide masses. [Now] you’ve identified Protein B, you’ve identified C, you’ve identified D, and you’ve identified E and you didn’t know what those proteins were before you did the experiment.
What does the process of X-ray crystallography do?
X-ray crystallography [provides] a precise three-dimensional view of a protein by shining beams of x-rays onto the protein [crystal] and then being able to solve the pattern of diffraction where those beams come off of the protein [crystal]. What you’re really trying to do in that procedure is to get a very precise three-dimensional view of a protein.
X-ray crystallography [gives] us this structure now of many, many different proteins at a very precise level, at the level of angstroms of tens of billions of a meter resolution, and we didn’t have that before. Ten, twenty, thirty years ago we had just a very few known protein structures. Now we have many, many structures that we have solved.
Can you explain how protein markers could be used to identify cancer?
In many cancer studies now — and I think this will extend to many different kinds of diseases-you’d like to be able to take a sample (typically of blood) from an individual and [ask if] something gone wrong in a given tissue [is] indicative of cancer. This may be true for heart disease [and for] many other diseases. What’s often gone wrong in a cancer is that the cell is now making proteins that it didn’t make before. It’s making more of them or it’s making different variants of them.
The example people often use is prostate cancer and what they see in men is prostate-specific antigen-the rise of that antigen, that protein, [indicates that] there’s a likelihood of prostrate cancer and further studies are done.
So you’d like to be able to look at ovarian cancer and other cancers and say that at an early stage we can [test] blood and say that there’s likely to be a problem here. We want to be able to find these proteins that are markers not just for the presence of the disease but the course of the disease, and how treatable it might be with different drugs.
There are many genetic changes that go on in a cancer cell vs. a normal cell and there are rearrangements, there are mutations-many of those changes are reflected in the fact that the cell makes different proteins than the normal equivalent. And so in all of those cases, one of the ways you might detect the presence of a cancer is the fact that different proteins are being made.
In previous days of trying to do this kind of study, you couldn’t look at all the proteins at once. [You would] make an educated guess [to find] the protein that varies in ovarian cancer and you could design an assay typically with an antibody to that particular protein and [see if] that protein change. Does it increase or decrease or does something else happen when an individual gets ovarian cancer?
But now you don’t have to limit yourself to a single protein. You can look at all the proteins and find any protein that might be a marker.
How has the ability to look at all the proteins at once changed medicine?
Proteomics is only slowly now having an impact on medicine and it’s likely that that impact is going to get greater and greater as the years go by. Ultimately I think we will be able to take tissue or blood samples and be able to diagnose at early stages a host of different diseases because of the pattern of changes in proteins. But that is not being done right now so I’d say the impact on medicine right now is not huge and it’s just beginning to escalate. I think the likely outcome is that [there] will be enormous consequences in the next five or ten years.
What is a scenario in the future for medicine?
What a lot of scientists and particularly now, biotechnology and pharmaceutical companies would like to be able to do is to take a profile of your proteins once a year and [be able to say]: “we’ve just noticed that particular rise in the level of a certain protein indicates that you’re in the early stages perhaps of a neurodegenerative disease or arthritis or diabetes or cancer and we might be able to take steps early on to treat you for that before it becomes a serious problem.” Proteomics is putting us in the position where we should be able to do that, not today but at some time in the future.
Can you explain why some plants have more genes than humans?
When the draft of the human sequence was first analyzed and published it was a surprise to many people that humans only had a few tens of thousands of genes, that we didn’t have the 100,000 or 150,000 that had been predicted. When the number of genes in humans was compared to simpler organisms, it was not that much greater even than a fruit fly or a nematode worm; it might be less than certain plants and other animals that people have analyzed.
There was this expectation perhaps that humans should have a lot more genes [because] we’re a lot more complicated than all these other organisms. One of the things that was found is that human genes are predicting proteins that are fairly complicated, that is they may occur in multiple different modules and different modules can carry out different functions. So this kind of complex arrangement of the proteins gives a lot of complexity to the cell.
Secondly, there’s this issue that each gene can make many different variants of proteins and so 30,000 or 40,000 genes still might be enough to make 300,000 or 400,000 different varieties of proteins.
But another big issue is that a lot of the complexity, be it of humans or any other organism, will come about through regulation. We can take existing genes and regulate them in new fashions [and] that introduces complexity. So it really doesn’t take that many genes to build something very complicated. The organism has the ability to turn on different genes at different times and that contributes to complexity.
How does the human proteome differ from that of other organisms?
What’s the difference between our proteins and say the proteins of a bacterium or a yeast cell that’s a single-celled organism? In fact, many of the proteins look virtually identical — you would hardly know when you were looking at the sequence of that protein whether it was the human protein or the bacterial protein. And these are proteins that have conserved structures because the function they carry out in the cell has not varied in maybe a billion years or more so the sequence hasn’t varied very much.
In other cases, there are human proteins that have multiple different domains and a highly complex structure, and you don’t see that same structure present within proteins from bacteria or yeast cells. There is a certain increasing complexity as you go through the evolution that contributes to the complexity of the structures you see.
What role do protein interactions play in complexity?
Protein interactions are what underpin all of these complex machines that carry out the different functions of a cell. The analogy that people often make is that of a three-dimensional jigsaw puzzle. You’re trying to slot one piece into an adjacent piece to another piece and the same thing is true in protein structures. Proteins must come together in highly complex arrangements where one protein fits into a complementary surface on another protein; then a third protein slots in and a four protein slots in.
But if you think of putting together a jigsaw puzzle, you’re hoping that the accuracy of the manufacturer is to maybe a fraction of an inch or so, so that the pieces come together. But if you think of protein structures, they have to be accurate to the level of angstroms-that is a ten-billionth of a meter — in order for those proteins to slot into each other, and if they aren’t that accurate then the complex can’t assemble.
How does this specificity affect protein interaction?
As proteins evolved over time, they evolved more and more interactions with proteins, and not just with other proteins but they also bind to DNA, RNA, lipids, and sugars-all kinds of structure in the cell. When that happens, a part of the protein will evolve to “see” another protein, and then a different part of the protein may evolve to “see” a third protein and then a fourth protein. And so over time, the protein is maintaining the first level of interaction-A with B and A with C-and then a different part of the protein is evolving so that now it recognizes D. And conversely, D is out there and maybe it didn’t bind to A. It bound to Protein F and G and D is now evolving so it still binds to F and G but now it can bind to A.
In doing so, you can [now] build up very large structures [that] can carry out very complicated tasks. Another thing that can happen is a protein can work in multiple different structures because in one case, Protein A binds to B, C, D and it carries out some function in DNA replication, and in another case Protein A is bound to Proteins K, L, M, N and is carrying out some process in DNA repair.
So proteins are not only capable of having lots of different partners, but they can be mutually exclusive. At one time it binds to one set of partners and another time it binds to a different set and that’s because those two different sets of proteins carry out two very different tasks in the cell.
Is that why humans only have tens of thousands but are still quite complex?
Part of the explanation for complexity can come about through a protein being able to have multiple different modules and those different modules are domains [that] interact with many other proteins and other components in the cell. So by building in more and more different modules within the same protein, evolution is capable of assembling very complicated structures. So at least part of the answer for why it doesn’t take that many genes to make a very complex looking cell is that proteins can assemble in different ways.
Can you discuss how techniques in proteomics have affected drug discovery?
In drug discovery, one of the features you’re often interested in is how a small molecule can slot into a particular protein and to do that you need the structure of that protein that the drug is binding to.
Proteomics allows a drug company or a biotechnology company to have lots and lots of different structures of proteins to work with. Instead of having to target a drug to only a single protein because that’s the only protein you had a structure for, you [now] have structures for tens or hundreds or even thousands of proteins [and] you’re able to consider any of them as potential drug targets.
Another issue that comes up in drug discovery is that when you add a drug to a cell or any organism, lots of changes occur that are not only occurring at the protein that you’re targeting. Other proteins may be affected, and if you’re able to look at what’s changing at a much broader level, then you may find that there are all kinds of side effects that make that an unworkable drug. You can learn that because you can look at so many more features of the cell than you could before.
We might be able to take cell lines or small animal models for a drug and be able to obtain enough information to know that a particular drug early on is going to be harmful without having to go to human testing. We can add a drug to a particular cell [and determine] what genes are affected, what genes turn on and which genes turn off and we have the ability to do that now for tens of thousands of genes.
And so we may find that when a particular drug is given to a cell line, that the pattern is indicative of kidney damage or liver damage and that that’s not going be a useful drug.
On the other hand, I think the issue that the drug industry has to face is that we may find that we can identify small fractions of the population with a particular disease that would be helped by a drug, even though the majority of the people who have that disease are not being helped. And if you stratify the population more and more so that lots of different diseases have lots of different subpopulations, each which can be helped by particular drugs, now the total medical cost goes skyrocketing because instead of having drugs that are generic that help most people with the disease they only help a small fraction. So you have [many different drugs] and the cost of developing and testing each of those drugs is enormously expensive. I think it’s a double-edged sword between being useful to save a lot of time and potentially eating up a huge amount of the total healthcare budget.
What are some other challenges of new drug design?
It’s often said that finding the target is not the challenge. That targets are proteins and we have the genomic structure for humans, so we know what most of the genes are by prediction and so we know what a lot of the proteins are, so finding the target may not be the issue. [However, you must find] the appropriate target amongst thousands of potential proteins that would be the right one for treating a disease. Before we had all of the genomic information, we didn’t know what a lot of these proteins were so we couldn’t even use them as drug targets because we didn’t know about them.
How do you determine what would bind to a target protein?
When scientists take protein structures now, they’re able to look at the level of angstroms of structure, so they can look for a pocket or a grove within a protein that a drug might slot into. They can design a particular drug computationally, and then [they] computationally dock a molecule that they’ve designed into the structure [to] see if that small molecule actually fits in. And it if doesn’t fit in, they can tinker with it, again still on the computer, and ask, if we had a methyl group here or we add another atom here, maybe that particular drug will fit better into the molecule. They can do all of this by design in the computer and then go ahead and synthesize the drug and then ask [if] that drug now really binds to the molecule the way they would have predicted-sometimes that works very well and they can come up with a good drug.
How was this done in the past?
Previously, most of it was done by a random process where you just made lots of different chemicals and just hoped that one of them would fit into your protein. That’s still being done today through either natural products or chemical synthesis. A lot of effort is still going into making tens of thousands of molecules and then seeing if any of them act as any good drugs.
In fact, I think of almost all the drugs out there, virtually all of them were found by this random screening method and very few have come about by rational design where scientists have looked at the structure and then looked at the chemistry and have made a marriage of those two.
Also, in the past, you would often use animal studies and find that when you give a drug to a mouse or some other animal there are side effects that make the drug unworkable. But [it] is a very expensive process to carry it to that stage, and if you can do the same kinds of studies by using markers earlier on (i.e., when you add a drug to a culture of cells and see certain changes occurring that are indicative of [harmful] side effects) then you can save yourself a lot of money.
What are some of the unanswered questions in this field?
I think that what the field of proteomics is very good at doing right now is to be able to identify what proteins are present in a sample. Where a lot of the action in the field is clearly moving toward is to do more than just identify a protein, but to be able to look at protein dynamics, to be able to say that the half-life of that protein has changed or that the protein has been modified in a certain way in response to a hormone or in response to a growth factor or in response to the cell going from a normal cell to a tumor cell.
And alternatively, you might find that a protein changes its location when you add a certain stimulus to the cell and so it’s these dynamic changes in protein abundances, modifications, localizations, and interactions. That’s what I think we’re moving towards as we get a better understanding of what proteins do and what they do on the large scale of thousands of proteins.
I’ve been a big proponent of developing technologies for proteomics and as you develop a new technology you suddenly are confronted with data of a type that you’ve never seen before and it allows you to think and dream about new possibilities.
Proteomics particularly is a field that will benefit enormously from new technologies that continue to develop and I don’t think anyone can foresee what those technologies are going to be. Each technology when it comes along will suddenly open up a huge area and people will say now we can analyze data sets that we couldn’t dream of before.
You mentioned new technologies: one of those new techniques that you developed was the yeast two-hybrid system. What are basics of this system?
Yeast two-hybrid is a method to identify and analyze protein-protein interactions. If you have a particular protein that you’re studying, you want to know all the other proteins [which it binds to or interacts with] and the two-hybrid method allows you to do this.
How the method works can be thought of as follows: If you think of a protein interaction as the equivalent of pieces of a jigsaw puzzle coming together, then if you have a box with tens of thousands of pieces of a jigsaw puzzle and you’re holding in your hand one piece of the jigsaw puzzle what you’d like to know is which of the pieces in that box fits exactly into the piece that you’re holding.
Two hybrid works as if you took the piece that you’re holding in your hand and you put a small battery on that piece with some wires hanging off. Then you went into the box and to every single piece of the thousands of pieces in the box you added a small light bulb. And now you take this piece with the battery and you throw it into the box and you shake it around-you’re looking for one piece to turn on the light of that light bulb. What that light tells you is that there’s been a specific interaction of two puzzle pieces; they’ve come together exactly in a way to position the wires and the battery next to the light bulb to turn on the light bulb. So when you see that light bulb go on you know you’ve got a puzzle interaction.
Two-hybrid does the same thing. It takes a particular protein that is a transcription factor which has two different functions. One function is to bind to DNA and the second function is to recruit one of these big protein machines that turns on a gene. Two-hybrid takes those two domains of the transcription factor and separates them so they no longer work because they’re not close enough to each other. The way they get close enough to each other is that each piece gets attached to two other proteins (the protein that you’re searching with and the protein you hope is the companion that matches up to that protein) and when the two proteins come together, you now get back transcription and a gene turns on and cells are able to grow. What you’ve effectively done is to uncouple what you’re looking for, and the way you’ve done that is by is by separating out this transcription factor and only bringing it back together when a protein-protein interaction occurs.
Will be ever map human proteome?
I’m not sure that you can ever say that you’ve reached a point where you understand the proteome because to me understanding the proteome or the function and identification and modification and interaction of every protein is very much as the same as saying we understand biology. Proteins carry out virtually all of the processes that we study in biology and if you knew everything about every protein I think we’d know everything that we want to know about biology.
So unlike the human genome where once we sequenced it from one end to the other you can say now we’re done, I think at the level of the proteome we don’t really have the whole proteome accomplished until we know everything.
Now if you if you simplify that problem to say that what we need to know is the parts list of all the proteins, then I think we can achieve that in a reasonable timeframe. We [will be able to] say, a given cell has this list of proteins in it. But if you extend proteomics to consider all of the other things that proteins can do, particularly their function then it’s a very difficult problem to say that we’re going to solve proteomics.
Why is this an exciting time to be in biology?
I think biologists now are poised to understand questions at a level of detail and understanding that we’ve never had before and to be able to do this for so many different organisms, so many different proteins and to integrate so many different types of information, and that’s a really exciting time. This will have implications for human medicine, for studying human evolution, for studying all kinds of fundamental processes in biology.