





Rediscovering Biology: Molecular to Global Perspectives
Online Textbook and Video
This online textbook chapter supports and extends the content of the Proteins and Proteomics video. The chapter covers protein modification, and tools for identifying protein structure and composition.
1. What Is Proteomics?
Unit 2 / Section 1
“If DNA is the genetic blueprint then what is the proteome? What are the proteins of the cell? The proteins of the cell are the walls, the floor, the plumbing, the beds, the furniture, the sinks, the glasses – everything that goes on in the house. All of those processes are being carried out by proteins and so DNA may be providing the instructions but all the work is really being done by the proteins.”
– Stanley Fields
What Is Proteomics?
A bacterial cell may seem simple but it’s actually a complex structure — a gel-like matrix of the cytoplasm, surrounded by both a lipid bilayer cell membrane and a cell wall. The cell must perform many functions including the intake of nutrients, the metabolism of those nutrients, growth, cell division, and the excretion of wastes. What molecules are involved? Although the cytoplasm contains water, proteins, carbohydrates, various ions, and assorted other molecules, proteins do most of the work. A typical bacterium requires more than 4,000 proteins for growth and reproduction. Not all of the proteins are made at the same time and some are made only under special conditions, such as when the cell is stressed or finds itself in a novel environment. The complement of proteins found in this single cell in a particular environment is the proteome. Proteomics is the study of the composition, structure, function, and interactions of the proteins directing the activities of each living cell.
If a bacterial cell needs more than 4,000 proteins, how many can we expect to find in animals? Mammals, including humans, have probably more than 100,000 proteins. Although the genome contains the genetic blueprint for an organism, the proteins of eukaryotes provide the unique structure and function that defines a particular cell or a tissue type, and ultimately defines an organism. Different types of cells make different proteins, so the proteome of one cell will be different from the proteome of another. In addition, cells that result from a disease, such as cancer, have a different proteome than normal cells. Therefore, understanding the normal proteome of a cell is critical in understanding the changes that occur as a result of disease. This knowledge can lead to an understanding of the molecular basis for the disease, which can then be used to develop treatment strategies. Knowing how the proteome changes as the organism grows may also provide insight into the mechanisms of development in healthy organisms.
Under the classical concept of “one gene makes one enzyme,” the proteome would simply comprise the products of all the genes present in the genome of an organism. But it is not that simple. The number of genes identified in the human genome is only about 30,000-35,000. How can only 35,000 genes encode more than 100,000 proteins? There are several possible answers to this question, which will be discussed in more detail below. One answer is that each gene may encode several proteins in a process called alternative splicing. Alternative splicing means that one gene may make different mRNA products and, hence, different proteins. Another answer is that one protein may be modified chemically after it is synthesized so that it acquires a different function. A third answer is that proteins interact with each other in complex pathways, and networks of pathways that may change their function. So, one gene may produce several, functionally different proteins in a variety of ways.
2. Introduction to Protein Structure
Unit 2 / Section 2

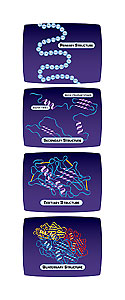
Figure 1. (Detail below)
Although proteins are unique, they share certain common characteristics
(Fig. 1). The primary structure of each protein is determined by the sequence of specific amino acids, encoded by the mRNA, which directs the proper folding of the polypeptide chain into the secondary structure. One type of secondary structure is the alpha helix, a region of the polypeptide that folds into a corkscrew shape. Beta strands are linear structures of polypeptides, bonding
together to form a flat beta-sheet. Other regions of secondary structure may include turns and random coils. These helices, strands, turns, and coils interact chemically with each other to form the unique three-dimensional shape of the protein, called the tertiary structure. For some proteins, a single polypeptide chain folded in its proper three-dimensional structure creates the final protein. Many proteins, however, have several different polypeptide subunits that make the final active protein. For these proteins, the interactions between the different subunits form the quaternary structure.
Discrete portions of proteins can fold independently from the rest of the protein and have their own function. These are called domains, and serve as one of the building blocks of that protein. Domains are evolutionarily mobile, capable of rearranging as new proteins evolve. There are thousands of structural domains, and many of them have been conserved widely across proteins. New proteins appear to have arisen over evolutionary time by bringing different domains together in a process known as domain shuffling. Domains often contain smaller motifs, consisting of a conserved pattern of amino acids, or of combinations of structural elements formed by the folding of nearby amino acid sequences. An example of a motif is a helix-loop-helix, which binds to DNA. Very similar motifs are found in many proteins that are not related. Scientists have classified conserved domains and motifs in a number of databases so that new proteins can be easily analyzed for the presence of these elements.
Figure 1. Detail: Primary Structure: The specific sequence of amino acids in a polypeptide chain. Secondary Structure: The folding of the polypeptide chain into specific shapes, such as the alpha-helix and beta-pleated sheet. Other regions of secondary structure may include turns and random coils. Tertiary Structure: The unique three-dimensional shape is the result of chemical interactions between amino acids that fold the regions of secondary structure. Quaternary Structure: The specific interaction of two or more polypeptide subunits.
3. Determining Protein Structure
Unit 2 / Section 3
While determining the polypeptide sequence resulting from gene translation is straightforward, determining the actual three-dimensional (3D) structure requires some sophisticated experimental techniques. One such long-standing technique is X-ray crystallography, which is based on the scattering of X-rays by the electrons in the crystal’s atoms. Think of the regular structure of table salt crystals. The atoms forming that structure are spaced very precisely in the crystal. Due to this regular spacing, a particular diffraction pattern forms when X-rays strike it. One can reconstruct the position of each atom in the crystal by observing the diffraction pattern and, thus, can make a three-dimensional map of the molecule. Although proteins are much more complex than table salt, researchers have crystallized many of them in their native configuration and have used X-ray crystallography to find their 3D structures. The 3D structures of proteins are available to all scientists in a public database called the “Protein Data Bank.”
Not all proteins can be crystallized, however. For example, membrane proteins have many hydrophobic amino acids and are particularly difficult to crystallize. A different technique to analyze proteins in solution is nuclear magnetic resonance (NMR). NMR is based on the principle that the nuclei of some elements’ atoms, such as hydrogen, resonate when a molecule, such as protein, is placed in a powerful magnetic field. NMR measures chemical shifts of the atoms’ nuclei in the protein, which is dependent on nearby atoms and on their distances from each other. The signals that NMR produces are a set of distances between specific pairs of atoms. NMR data generate models of possible structures, rather than a single structure. For smaller proteins in particular, NMR can quite accurately predict the 3D structure.
Despite advances in techniques for determining protein structure, the structures of many proteins are still unknown. With the help of protein prediction programs, computer analysis of genome sequences is producing thousands of new hypothetical proteins of unknown structure and function. These proteins are called “hypothetical proteins” because they represent the products predicted from the gene sequence; however, there is, as yet, no evidence that they are actually made and there is no known function for them.
Computer programs may help determine the structure of proteins whose function is not yet known. By comparing the sequence of the unknown protein to proteins with known 3D structures, these programs can make a predictive model of the unknown protein’s structure using the known proteins as templates. The success of this method depends on the quality of the match between the known template proteins and the unknown target protein. In addition, when the function of the template protein is known, it may help identify the function of the unknown protein. These prediction programs do not produce structures with the detail or reliability of experimental techniques such as X-ray crystallography. They do, however, provide a means to analyze — in a reasonable time period — the large number of new proteins identified by the analysis of whole genomes.
4. Structure and Function Relationships of Proteins
Unit 2 / Section 4
The three-dimensional structure of a protein defines not only its size and shape, but also its function. One characteristic that affects function is the hydrophobicity of a protein, which is determined by the primary and secondary structure. For example, let’s look at membrane proteins. Membranes contain large amounts of lipids, which are notoriously hydrophobic (water and oil don’t mix). The membrane-spanning regions of membrane proteins are typically alpha-helices, made of hydrophobic amino acids. These hydrophobic regions interact favorably with the hydrophobic lipids in the membrane, forming stable membrane structures.
Hemoglobin is a soluble protein – found in the cytoplasm of red blood cells as single molecules – which bind oxygen and carry it to the tissues. In sickle cell anemia, a mutation in the beta-globin protein of the red blood cell increases its hydrophobicity and causes the mutant protein molecules to stick to each other, avoiding the aqueous environment. Chains of hemoglobin change the shape of the red blood cell from round to a sickle shape, which causes the cells to collect in narrow blood vessels.

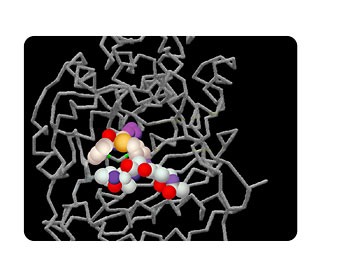
Figure 2. The active site of the penicillin-binding protein. The gray stick-like structures represent the secondary and tertiary structure of the penicillin-binding protein. Binding of the antibiotic, the substrate, to the active site blocks the normal action of the protein in the bacterial cell, resulting in death of the cell.
The folding of a protein allows for interactions between amino acids that may be distant from each other in the primary sequence of the protein. In enzymes, some of these amino acids form a site in the structure that catalyzes the enzymatic reaction. This site, called the active site of the enzyme, has amino acids that bind specifically to the substrate molecule, also called a ligand (Fig. 2). In a similar manner, certain sites in cell receptor proteins bind to specific ligand molecules that the receptor recognizes.
Alterations in amino acids that may be distant from each other in the primary sequence can lead to changes in folding. It may also cause changes in chemical interactions among amino acids at the active site, which alter the enzyme activity or binding of the ligands to receptor proteins. Binding of ligands to an active site requires specific amino acids. Therefore, an active site in a new enzyme that belongs to the same family as a known enzyme can usually be identified by its similarity to the active site of the known protein. Computer programs can use the information from a database of known enzymes to predict the active site of a new protein using a template-based method, similar to that described above for determining the three-dimensional structure of a protein. Once the program has identified the potential ligand-binding sites, other programs can test the fit and the binding ability of thousands of possible ligand molecules — even theoretical ligands that may not yet exist. This has tremendous possibilities for the design of new drugs, particularly for cancer therapy.
5. Protein Modification
Unit 2 / Section 5
The complexities of the 3D structure of proteins are not the only difficulty in characterizing proteins. Many proteins contain additional chemicals that modify their structure. The final structure of a protein may include any number of modifications that occur during and after the synthesis of the protein on the ribosome. These post-translational modifications change the size and structure of the final protein. Some modifications occur after a protein is made; others occur during translation of the protein and are required for proper folding of the protein. One possible modification is enzymatic cleavage of the original polypeptide by proteases to produce a smaller product. Other modifications include the addition of sugar molecules to certain amino acids in the protein (glycosylation), or the addition of a phosphate group (]tooltip]phosphorylation[/tooltip]) or a sulfate group (sulfation).
Many proteins are modified by proteases that remove short peptides from either end of the protein. The shortened polypeptides then fold into an active protein. One of the most common of these cleavages is the removal of specific signal peptides. These peptides target proteins for transport to a particular cellular organelle in a process known as protein sorting. An example of this is the hormone insulin, which is made as preproinsulin. After removal of the 24-amino-acid signal peptide from preproinsulin to form proinsulin, the latter polypeptide is further processed in the endoplasmic reticulum. This produces the final hormone, insulin, which is released from the cell.
Glycosylation — the addition of specific short-chain sugars to asparagine, serine, or threonine — is very common in membrane proteins that form structural components of the cell surface. These proteins, called glycoproteins, are important in many cell processes, including binding by receptors and eliciting an immune response. Glycoproteins are often specific cell markers. For example, ABO blood types result from the presence or absence of specific glycoproteins (A-type, B-type, both, or neither) on the surface of red blood cells. Human immunoglobulin G (IgG) is also a glycoprotein in which the sugar appears to very important for the normal function of the protein in the immune response. Scientists have discovered that abnormal sugars in IgG strongly correlate with the autoimmune disease called rheumatoid arthritis, characterized by chronic joint inflammation, and the presence of antibodies directed against IgG and other host proteins.
Reversible phosphorylation of threonine, serine, or tyrosine residues by enzymes called kinases (which add a phosphate) and phosphatases (which remove the phosphate) play an important role in the regulation of many cell processes, such as growth and cell cycle control. (See the Cell Biology and Cancer unit.) Phosphorylation may occur sequentially from one protein to another, resulting in a series of activations called a “phosphorylation cascade.”
6. Genomics-Based Predictions of Cellular Proteins
Unit 2 / Section 6
We now have large databases of gene sequences, predicted protein sequences, and known 3D protein structures, yet we still don’t know the total protein composition of a cell. Determining the proteome of a cell is a complicated task. There are two approaches to obtaining this information: computer-based and experimental.
The computer-based method uses the genome sequence of an organism to predict genes, based on known characteristics of protein-coding regions of the genome. (See the Genomics unit for a discussion of computer-based methods for gene identification and microarrays to identify expressed genes.) However, even if we know that a particular sequence is a gene, we don’t necessarily know all the possible proteins it makes.

Figure 3. More than one protein can be made from a gene. In this case, three different mRNA molecules are made from one gene. The exons (the numbered boxes) can combine in different configurations to eventually form different proteins.
One reason is that one gene may produce more than one mRNA. RNA splicing is the normal process in which intron sequences are removed from the pre-mRNA, producing the mRNA, which corresponds to the exons. However, some transcripts can be spliced in alternative ways (alternative splicing), joining different exons (Fig. 3). The result is two or more different mRNA molecules from one gene. Variants of a protein produced by alternative splicing may have similar physiological activity, a different and unrelated activity, or no activity at all. According to one estimate, about forty percent of human genes are alternatively spliced. This is one mechanism that accounts for the relatively large number of proteins produced by only about 35,000 human genes.
A more direct approach to identify proteins in a cell is to measure enzyme activities and other functions for which there are biochemical assays. In some cases, we can identify the function of new proteins by combining our knowledge of metabolic pathways in many organisms with the predicted function from genome analysis. With this type of information, researchers can readily identify new enzymes. To do this, they examine the similarity of the genome sequences to known enzymes, as well as the presence (in the same genome) of the proteins that are required for the other steps in the metabolic pathway.
7. 2D Gel Electrophoresis to Identify Cellular Proteins
Unit 2 / Section 7
While computer-based methods are powerful, they can only predict the function of proteins for which some information is already available. How do we understand the proteins that we don’t already know about? This requires experimental approaches.
One way to identify proteins is to extract all the proteins from a sample of cells and separate them in a gel matrix, using a technique called polyacrylamide gel electrophoresis (PAGE). The proteins are separated by size, with the smaller proteins moving faster through the gel than the larger proteins. After staining, a pattern of bands appears that corresponds to the proteins in the cell. However, this technique can only resolve a few hundred proteins, and cannot separate proteins of very similar size.

Figure 4. Haemophilus influenzae cell proteins separated by 2D gel electrophoresis. The basic proteins are to the right of the gel and the acidic proteins to the left. High molecular weight proteins are to the top of the gel.
A modification of this procedure – called 2D gel electrophoresis — separates proteins into two dimensions, using two different characteristics. Proteins are separated in the first dimension by their isoelectric point (pI), the specific point at which the net charge of the protein is zero. These separated proteins, in a flat gel strip, are then placed on a standard polyacrylamide gel. Every protein band that was separated in the first dimension according to its isoelectric point is now separated in the second dimension by its size. The result is small spots, each representing a protein; even proteins of the same size will be resolved if they have a different isoelectric point. A good 2D gel can resolve one thousand to two thousand proteins, which appear, after staining, as dots in the gel (Fig. 4). This technique is useful when comparing two similar samples to find specific protein differences; for example, comparing the proteins in a tumor cell versus a normal cell. However, it can miss very small proteins or non-abundant proteins.
8. Mass Spectrometry to Identify Cellular Proteins
Unit 2 / Section 8
While the 2D gel method easily separates proteins, it doesn’t identify them. If there are differences in spots between the proteins in a cancer cell and a normal cell, this method cannot determine the actual identity of the different proteins in the two cell types. To identify these proteins, individual spots are excised from 2D gels and then subjected to mass spectrometry, which separates charged particles, or ions, according to mass. First, the molecules in the sample are ionized to produce a population of charged molecules. A mass analyzer then separates the sample’s molecules based on their mass to charge ratio. A detector then produces a peak for each ion; this peak gives the mass and represents the amount of the ion. A computer program reads the complex spectral information from the mass spectrometry process. The program matches the information on each peptide’s mass against the mass of theoretical, predicted peptides, based on known proteins in databases. This is called peptide mass mapping. With many different peptides for each protein, the computer can match the sequence to one or more known proteins. Peptide mass mapping can only be used in situations where the genome has been sequenced and all predicted proteins for the genome are known.
Another application of mass spectrometry is protein fingerprinting. This technique has been used to identify unique sets of proteins in blood, which serve as markers for different forms of cancer. Interestingly, for this method to be useful, we do not need to know the actual identities of the particular proteins used as markers for disease. Instead, this technique relies on pattern recognition software. Using training data from samples from individuals with and without cancer, the program searches for a particular pattern of peaks that correlates with cancer. This technique requires only a drop of blood and does not require any detailed genetic information; however, its accuracy in predicting some forms of cancer is limited because the number of marker peptides is not sufficiently large. As more samples are evaluated, the accuracy will likely increase because the software will be able to find more accurate peptide patterns correlating to cancer. Proteomic fingerprinting holds great promise as a diagnostic tool for a variety of diseases that produce distinctive patterns of proteins in blood.
9. Identifying Protein Interactions
Unit 2 / Section 9
While it is convenient to think of proteins as discrete and independent molecules, this is actually an oversimplified view. Many proteins require other proteins or cofactors for activity; and proteins involved in signal transduction, protein trafficking, cell cycle, and gene regulation must interact with other proteins in those processes. Many of these interactions require particular domains called interaction domains. Proteins involved in the interactions contain combinations of interaction domains (for interaction with other proteins) and catalytic domains (for function of the protein). The interaction domain can bind the partner protein, even in the absence of the rest of the protein. Interaction domains are often quite versatile, capable of binding a variety of related ligands. In addition, one protein may contain several different interaction domains. The modular nature of these domains allows the protein to interact with multiple target proteins in the cell; thus it provides a mechanism for integration and control of information from protein to protein in a cell. Such protein-protein interactions form the basis for our current understanding of cell signaling pathways and protein networks that regulate all the activities in a cell.
Because protein-protein interactions regulate the activities of cells, identifying them is critical to understanding cellular processes. Mass spectrometry techniques have been developed for large-scale screening to identify interacting proteins. For example, hundreds of known proteins in yeast were engineered to contain a biochemical tag that would allow the tagged protein to be separated from other proteins in a cell extract. This was done gently so that other proteins bound to the tagged protein would still be attached. The tagged protein, along with any associated proteins, was then analyzed by mass spectrometry. The results revealed that about eighty-five percent of these proteins were associated with other proteins. Although most interacted with many other proteins, in some cases two different protein complexes had at least one protein in common. Among the most intriguing questions to come out of this research were what controls which proteins interact and — for those that interact in multiple complexes – how do these proteins know which complex to join?
10. The Yeast Two-Hybrid System
Unit 2 / Section 10
The yeast two-hybrid system is a powerful technique for identifying multiprotein complexes. Using genetically engineered yeast, scientists can identify complexes when specific pairs of interacting proteins activate expression of a reporter gene. One often-used reporter gene is the lacZ gene. When two proteins interact in the yeast cell they activate expression of this gene, allowing yeast cells to metabolize an indicator that turns these cells a different color. The interacting proteins are then identified from the colonies formed by these colored cells. The two-hybrid system has been expanded to use microarrays of cloned yeast genes (see below). These large-scale yeast two-hybrid assays can provide information on thousands of protein-protein interactions. Using this technology, researchers are identifying all the proteins in yeast that interact, and they will then map the complex network of cellular functions to these interacting proteins.
11. Protein Microarrays
Unit 2 / Section 11
Another strategy for the large-scale study of proteins is similar to the DNA microarrays, which measure gene expression in different cells types. (See the Genomics unit.) Based on the rapid, large-scale technology (often called high-throughput technology) that was developed for DNA microarrays, scientists have developed similar microarrays for proteins. In a protein microarray, very small amounts of different purified proteins are placed on a glass slide in a pattern of columns and rows. These proteins must be pure, fairly concentrated, and folded in their active state. Various types of probe molecules may be added to the array and assayed for ability to bind or react with the protein. Typically the probe molecules are labeled with a fluorescent dye, so that when the probe binds to the protein it results in a fluorescent signal that can be read by a laser scanner.
This technology can complement other techniques, such as mass spectrometry and yeast two-hybrid assays, to identify thousands of protein-protein interactions. Protein arrays can be screened for their ability to bind other proteins in a complex, receptors, antibodies; lipids; enzymes; peptides; hormones; specific DNA sequences; or small molecules, such as potential new drugs. One of the most promising applications for protein microarrays is the rapid detection or diagnosis of disease by identifying a set of proteins associated with the disease.
One example of the use of this technique is the development of a microarray that may help in the treatment of cancer. This microarray contains many different mutant forms of a protein called p53. P53 is an anti-cancer protein, called a “tumor-suppressor protein,” and about half of all cancers have mutations in p53 (See the Cell Biology and Cancer unit.). Researchers can screen the immobilized mutant p53 proteins in the microarray for biological activity, as well as for new drugs that can restore its normal tumor-suppressing function.
12. Protein Networks
Unit 2 / Section 12

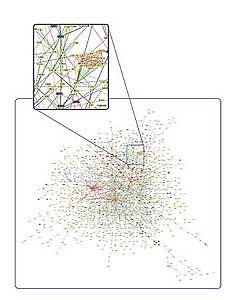
Figure 5. A network of protein-protein interactions in a yeast cell.
The cell is a complex and dynamic system of networks of interacting molecules.
An understanding of the cell requires analyzing these complex interactions as a system. Systems biology takes the approach that the powerful high-throughput techniques, developed as part of whole genome and proteome analysis, will allow the simultaneous study of complex interactions of networks of molecules, including DNA, RNA, and proteins. Fully understanding complex networks of molecular interactions in the cell require a combination of several different experimental techniques, including DNA and protein microarrays, mass spectral analysis, and two-hybrid analysis. This, combined with the power of computers to analyze the massive amount of data, produces models of interacting networks, which better describe the workings of a cell (Fig. 5).
Figure 5 credit: Schwikowski et al, A NETWORK OF PROTEIN-PROTEIN INTERACTIONS IN YEAST (2000). Courtesy of Nature Publishing Group.
13. Proteomes in Different Organisms
Unit 2 / Section 13
Although scientists have sequenced dozens of genomes from organisms as diverse as viruses, bacteria, nematode, fruit fly, pufferfish, mouse, and human, we still don’t know what uniquely characterizes each of these organisms. For example, both mouse and human genomes contain around 30,000 genes. How many of these genes do they share? Based on comparisons of the two genomes, ninety-nine percent of the genes are conserved in both species and are, thus, derived from a common evolutionary ancestor. The remaining one percent represents genes that evolved independently in mice or humans. If these two organisms share so many similar genes, how can they be so different? A simple example may help us to understand that the presence of a gene does not mean that the protein is expressed. Pigs produce cell surface proteins, which are modified by glycosylation to contain a sugar called galactose (GAL). Those GAL-proteins, present in pig blood vessels, are seen as foreign by the human immune system. This leads to the very rapid destruction of pig organs that have been transplanted into humans when a human organ was not available. Interestingly, humans lack GAL-proteins but still have the gene for making them; the gene is not expressed in humans. Therefore, the presence of a gene does not mean that it is expressed. In fact, every somatic cell in an organism shares the same genes; so, the differences between tissue types – say liver and heart – result from differences in gene expression. (See the Genetics of Development unit.)
Identification of proteins may provide the most useful information in determining the significant differences between species. How different are the proteins in even closely related organisms? With the development of proteomic techniques, scientists are beginning to tackle this difficult question. One answer is that very similar genes in two organisms may be expressed very differently. Dr. Svante Pääbo of the Max Planck Institute for Evolutionary Anthropology analyzed the proteins from the brains of humans and chimps. (See the Genomics and Human Evolution units.) He found that many, very similar genes produced much more protein in human brain cells than in chimp brain cells. In contrast, the same type of experiment done with blood or liver cells showed much less difference between humans and chimp in the amount of protein produced.
At a different level, there are some clear differences in protein composition between the cells of eukaryotes and those of the other kingdoms. One is that eukaryotes have many more long proteins, more proteins with regular secondary structure and less random globular structure, and more loop regions in their proteins. Certain conserved structural domains show up in proteins but are used in a number of different pathways. While there are many protein homologues conserved across many different organisms, some proteins are unique to one organism. As more genomes and proteomes are characterized, comparative genomics and proteomics will allow scientists to further understand how organisms differ.
14. Proteomics and Drug Discovery
Unit 2 / Section 14
One of the most promising developments to come from the study of human genes and proteins has been the identification of potential new drugs for the treatment of disease. This relies on genome and proteome information to identify proteins associated with a disease, which computer software can then use as targets for new drugs. For example, if a certain protein is implicated in a disease, the 3D structure of that protein provides the information a computer program needs to design drugs to interfere with the action of the protein. A molecule that fits the active site of an enzyme, but cannot be released by the enzyme, will inactivate the enzyme. This is the basis of new drug-discovery tools, which aim to find new drugs to inactivate proteins involved in disease. As genetic differences among individuals are found, researchers will use these same techniques to develop personalized drugs that are more effective for the individual.
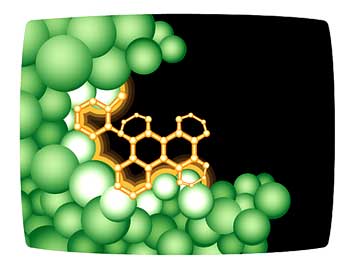
Virtual ligand screening is a computer technique that attempts to fit millions of small molecules to the three-dimensional structure of a protein.

Figure 6. In virtual ligand screening, the three-dimensional image of the protein is fed into a computer, which attempts to fit millions of small molecules to a targeted active site. Small molecules that bind well to the protein become good leads for potential new drugs.
The computer rates the quality of the fit to various sites in the protein, with the goal of either enhancing or disabling the function of the protein, depending on its function in the cell. A good example of this is the identification of new drugs to target and inactivate the HIV-1 protease. The HIV-1 protease is an enzyme that cleaves a very large HIV protein into smaller, functional proteins. The virus cannot survive without this enzyme; therefore, it is one of the most effective protein targets for killing HIV
(Fig. 6).
Because many proteins have multiple functions, it may be necessary to develop drugs for each function of a multitask protein. In addition, most proteins act as part of complexes and networks, which may also affect the way a protein acts in a cell. This may also affect the ability of drugs to disable the protein. Understanding the proteome, the structure, and function of each protein, and the complexities of protein-protein interactions will be critical for developing the most effective diagnostic techniques and disease treatments in the future.
15. Ethics and the Economics of Drug Discovery
Unit 2 / Section 15
Drug discovery is simple compared to drug development, which requires testing the efficacy and the safety of new drugs through clinical trials. The time (twelve to fifteen years) and cost (approximately 800 million dollars) of drug development are significant economic factors that limit the number of new drugs that come to market; many approved drugs never recover the cost of their development. How do companies decide which promising new drugs to develop? Clearly, there must be very good evidence that the new drug will be effective. But that is not enough; companies also carefully consider the economics of each potential new drug. What is the size of the market for that new drug? How strong is the demand? How effective are current drugs and what are their costs?
The harsh reality of these economics is that new drugs that may benefit only a few are unlikely to make it to clinical trials. Drugs that may benefit millions of people in developing countries too poor to pay for the new drug will also have a low priority for development. While AIDS, malaria, and tuberculosis affect countries that together contain ninety percent of the world’s population, only about ten percent of the world’s medical research funding is targeted at these diseases. Partnerships among government agencies, charitable organizations, and the pharmaceutical industry may allow companies to allocate some of their resources to developing drugs that will never recover their costs. In 2001 GlaxoSmithKline Biologicals, in partnership with the World Health Organization and the non-profit organization Program for Appropriate Technology in Health, began a program to develop a vaccine for childhood malaria.
Many currently patented drugs could be manufactured in third world countries as generic versions. However, pharmaceutical companies have strongly opposed this practice, fearing that these generic drugs will be inferior to the name brands and would enter the U.S. and European markets at low prices. Brazil has registered generic versions of several AIDS drugs and manufactures them for itself and other developing countries. In response to worldwide pressure, drug companies have agreed to sell some AIDS drugs at deep discounts to developing countries. However, even with the discounts, the price is much higher than the generic version, limiting the number of AIDS victims who can be treated in poorer nations.