








Rediscovering Biology: Molecular to Global Perspectives
Evolution and Phylogenetics Expert Interview Transcripts: Timothy Read
 Assistant Researcher
Assistant Researcher
Read, is an assistant researcher with the Microbial Genetics Group at The Institute for Genomic Research in Maryland. In addition to examining the genome and evolutionary history of Bacillus anthracis, the bacterium that causes anthrax, Read also explores microbial genes as potential drug targets.
Interview Transcript
Read is an assistant researcher with the Microbial Genetics Group at The Institute for Genomic Research in Maryland. In addition to examining the genome and evolutionary history of Bacillus anthracis, the bacterium that causes anthrax, Read also explores microbial genes as potential drug targets.
How important is molecular data in proposing evolutionary relationships?
Well, I think you have to base evolutionary relationships on the DNA sequence. It’s the most fundamental way of classifying organisms. Any other type of data essentially represents some sort of phenotype that relates back to the DNA sequence.
How has molecular data changed the way that evolution is studied?
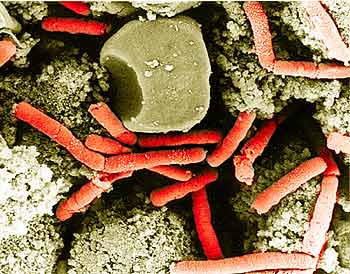
The original people who built phylogenies, trees, could only work with what they had available, which was essentially morphological differences, and observable characteristics. Now that we have DNA sequences we can really get to the root of evolution. One obvious example is bacteria because within bacteria there’s an enormous genetic variation in the DNA. But when you look at their morphology they actually look very similar. They’re very small. They’re typically either round or rod-shaped, either Gram-positive or Gram-negative. There are very few features you can really use to distinguish them just by looking at them under a microscope. But when you actually look at their DNA sequences, you see a lot of variation.
Can you explain how that research would be accomplished using DNA?
A typical thing to do would be to take a DNA sequence of a gene that you know is conserved in all organisms-something very fundamental. A good choice would be the gene for a ribosomal protein. You would compare the sequence of, say, anthrax to the same gene in many other bacteria. You can even compare them to eukaryotic cells like humans or yeast. Then you would try to make a tree based on the sequences, the proteins that are most similar. So anthrax would be on a branch of the tree with a closely related bacteria because the sequences of the genes are very similar.
Can you describe your recent work with Bacillus anthracis?
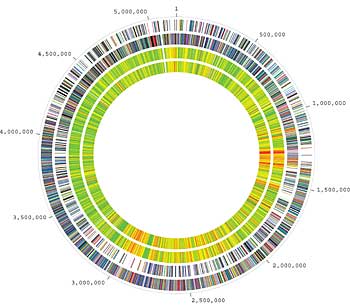
We started sequencing Bacillus anthracis a while ago; the reasons for sequencing the bacteria really have to do with the shift in the way biology is done. It’s increasingly important that you characterize an interesting organism by knowing its entire genome sequence. And a lot of bacteria that cause disease have had their genome sequenced, like salmonella and Escherichia coli. And anthrax was another bacteria that causes disease, so it was important to get its genome sequence.
Obviously, after the bioterror attacks [of 2001], there was even more increased interest in anthrax because it’s an effective biological warfare agent. And the project that we took on was to sequence the genome of the anthrax strain used in the Florida attack and compare it to the genome that we were sequencing before that occurred.
And what did you find in those experiments?
We knew that the genome we were sequencing before and the genome from the Florida attack were, in fact, almost identical, because essentially they’d been both distributed from the same source lab just a few years ago. So this was a very unusual use of genomics, to essentially find the tiny differences between what many people would consider identical organisms. But by using some tools for comparing sequences and looking back in our actual raw data, we could find measurable differences between the two organisms.
And what did those differences tell you about the anthrax used in the attacks?
The reason the genomes of the two strains we were sequencing were considered identical was based on a genotypic analysis, essentially like a DNA fingerprint. And this was based on analysis of the VNTR’s, or Variable Number of Tandem Repeats, which essentially looks at portions of the genome that were known to be highly variable.
Essentially, these VNTR’s are small repeats of DNA sequence that exist in multiple copies, and the number of these repeats can vary. And these regions we studied were known to be highly variable parts of the genome, and essentially the two strains we were sequencing were identical in the pattern of their VNTR’s, and that’s why they’re considered identical.
There is, however, another level of variation that’s not associated with highly variable portions of the genome, and these can be small deletions or they can be Single Nucleotide Polymorphisms, which we call SNP’s (“snips”), which occur by random mutation. It’s tricky to find them because they could be anywhere in the genome. There’s no way to predict where they’re going to occur, but by comparing the complete genomes of the two organisms we could find these very rare, unpredictable differences.
Did you find any evidence of genetic engineering in the anthrax strain?
Because we’d already sequenced one organism, we had an idea of the gene content and what genes we would find. Then, in studying the terrorist strain, one of the hypotheses was that someone had inserted something into the Florida sequence to make it more virulent. So we did a search of all the sequences to make sure that they matched the original genomic sequence. And basically, we found no evidence that there were genes inserted into the Florida strain compared to what we’d been seeing before.
And so you constructed a phylogeny to compare the genomic differences?
First of all, in this situation, we found so few differences. And the differences we saw were SNP’s and VNTR’s, which are really quite two different things. So we made a sort of phylogeny that you might create for multiple gene comparison in different species, for example. We were really just trying to construct what we thought was maybe the closest related strains based on some hypotheses. Comparing SNP’s is probably the best way to determine what we think are the most closely related strains and what are the furthest apart. But there were so few SNP’s that were different between the strains that it was hard to use those.
What was the role of phylogeny in analyzing the sequence?
There are some really interesting questions that we want to look at here. Firstly, the sequence was used to find the VNTR’s, to create a genetic fingerprint. We then classified the anthrax strains into different groups based on this fingerprint. So now we’re actually sequencing representative genomes from these different groups. And what we want to find essentially are SNP’s from these different groups that we can go back, test lots of anthrax strains, and see if the phylogeny we build on the basis of the SNP’s matches the phylogeny that was built based on the VNTR’s, and I think that’ll be very interesting.
Also, we are going to sequence very closely related neighbors of Bacillus anthracis, and what I hope will happen is that some of the differences we find in the SNP’s in anthrax will be shared with these new neighbors. And if that’s the case, we can actually root the tree. We can see which of the Bacillus anthracis strains are more closely related to the ancestor, and thus we can tell which evolved first and which are newest. Maybe we’ll be able to trace the path of how anthrax evolved, where it came from, and how quickly it’s been moving around.
What other further applications might this research lead to?
For other pathogens, I think similar research is going on. Anthrax is really quite a unique situation because the different strains, from all over the world, are almost identical in sequence. You don’t normally see that, even in very closely related pathogens like Mycobacterium tuberculosis and Yersinia pestis, which causes plague.
In terms of evolution, I think the first thing is tracing the pathway. The second is to see how it has evolved by seeing which genes are being changed as it evolves. So essentially it’s telling us where there’s selection for anthrax to change and how these genes might be very important in how anthrax causes disease.
Can you describe functional genomics?
If you start with a genome sequence, you can use that information to really tackle the biology of the organism. There are two basic approaches to take. One is called a microarray, where you take all the genes you found using the genomic method, which is about 6,000 in the case of anthrax, and you essentially create a gene chip where you actually print these genes on to a glass slide, and then you present the chip with the mRNA that anthrax has generated, which represents the proteins that it expresses. And then you can see which genes will hybridize with anthrax RNA message, which tells you which genes are expressed under certain conditions. And an obvious thing to look at for anthrax is maybe when it’s causing disease, or are virulent, which genes are producing mRNA, which genes are being “turned on,” being expressed. That’s one functional genomics approach.
Another functional genomics approach we’re working with is called “proteomics,” and the idea is you take the entire anthrax cell and you can identify which of the 6,000 proteins are being expressed in that cell at that time, under any specific conditions. So the proteomic and the microarray approaches to genomics are sort of linked together to give us this story about how anthrax causes disease.
How can you go about identifying these rare regions of anthrax DNA variability?
You can find regions that you think will be different using just one genome by identifying the VNTR’s. In one genome, you might find, say, twenty of these repeat units and then you think to yourself, well, in other genomes this will be a highly mutable area; this number will probably change. But the SNP’s, these are less obvious and predictable in terms of where they are, and you find them by comparing the genome sequence of one organism against another.
What are the implications of the work, and what do you hope to learn from it?
Obviously, one of the implications is for forensics, as we’ve shown, if you’re really interested in an organism for any reason-and obviously a bioterror attack you want attribution, you want to try and find where this organism comes from. Genome sequencing is also useful for developing what they call “signatures.” A lot of people want to be able to detect anthrax in the environment, and they can use the genome sequence by comparing it to closer related organisms to find the unique parts of the anthrax DNA. So a DNA signature is something that you only find in anthrax and nothing else, and you can use that to help detect it. You can also use the DNA sequence to find vaccine candidates, to predict the type of molecule that might be on the surface of anthrax. And the same sort of technique is useful for designing new drugs. We can look at the genome sequence and we can say, this protein I can design a drug and it will kill the cell if it hits this protein.
What other techniques will be useful?
Essentially, bioinformatics is extremely important in what we do, and this is to really look at genome sequences and apply computational analysis to them. And it’s hard to find small differences, real differences, between very large sequences because there’s always a certain background of a sort, a noise, differences that aren’t real, so finding real differences using computational methods is a real breakthrough.
Have you had any surprises along the way?
I think the fact that you can use something like a whole genome sequence just to find one or two differences was surprising. I guess it’s hard to emphasize that too much. That was something that we thought might work and it did work. And I think that’s been picked up very quickly as a valuable tool, for forensics so that’s a real surprise.
What about the approach will be different from genotyping?
Genotyping will carry on because essentially a way to identify the portions of the genome that are the most variable and you use that to genotype organisms. It’s a very efficient, quick way of screening lots of strains cheaply so that will definitely still play a role. And you can use SNPs for genotyping, too, so you can use the data from these genome sequence comparisons. You might find say a thousand SNP’s but you might make up your genotyping scheme from say twenty that just distinguish strains into the sort of branches of the tree rather than the individual twigs.
So can molecular systematics work as a weapon against biological terrorism?
It already is. It’s already a major part of the preparation. Molecular systematics is a major part of the war against bioterrorism.
Does the change in the patterns of nucleotide sequences show evolution?
That’s what evolution is: a change in the nucleotide sequence.
What differences might exist between phylogenies that are based on genomes vs. other molecular data?
Well, I don’t think anyone’s actually used entire genomes because it’s quite difficult to do that. What they have done is to use single genes or groups of genes, because when you have entire genomes-say you have 6,000 genes in anthrax-maybe half of those genes won’t be found in E. coli. So if you use an entire genome you’re not comparing like to like. So one of the important things in constructing phylogenies is to compare a few highly conserved genes across a wide group of organisms.
The alternative to using genes-and I don’t want to oversimplify things-is to use protein sequences. The protein is obviously encoded by the gene, but essentially over the course of evolutionary time, there’s more change in the nucleotide sequence of the gene than there is in the final protein that’s encoded by the gene. So really when people are looking over wide evolutionary distances they’re looking at protein sequences rather than genes because the genetic code has just changed too much to really get anything meaningful out of it.
So in the example, I gave previously, the ribosomal protein we chose, you’d use the protein amino acid sequence encoded by that gene to compare bacteria to one another rather than the nucleotide sequence because the protein sequence will be more meaningful.
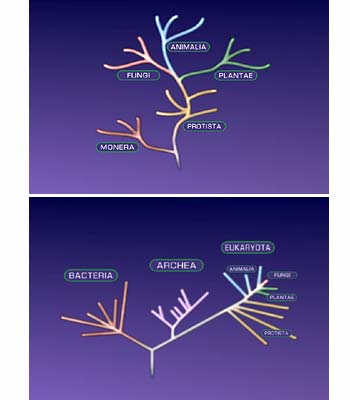
How does this method of creating a phylogeny relate to the rRNA tree of life?
Using a genomic approach, we now have many hundreds of genomes of bacteria. Maybe in ten years and that’ll be more like thousands, so this is obviously the future of phylogenetics. I think that the tree of life created [by Carl Woese] from the rRNA gene sequence, again, you’re just looking at one highly conserved gene, so you can do this for all organisms, you can ask those questions.
What big questions are you left with?
I’m intrigued, as you said, by anthrax because the bacteria has obviously evolved very recently, and it may interact with some of our ideas about history and anthropology, how it moved around the world and what’s happened since. The phylogeny of anthrax actually we may be able to relate to sociological things as well, which I think is fascinating. I’m also very interested in the method of evolution of anthrax and the story behind it. I think it may be an interesting story of how something that lives in soil and is quite versatile in how it lives. You know it can do quite a lot of different things. When it becomes a pathogen by just acquiring a couple of genes, a lot of its genes lose their roles and lose their meaning and so you have a lot of genome change in a short period of time as genes are sort of cut off and are lost. So I think the evolution of anthrax in that regard is also very interesting.
What would you say are the implications of your work for biology?
I would say what we’re doing now is something unique. No one’s really looked at organisms this closely related on the scale that we’re doing it. We’re comparing 14 genomes of an organism that many people call identical, and I think we’re going to find very few differences but they can be meaningful in that we’re really going to understand how evolution occurs in a short stretch of time. So I think it will be significant in that regard.
What would you want teachers or the public to understand about the field?
I think one thing is that science takes a lot of people to do it. And in fact, we’ve been talking about a big project with lots of people involved and a lot of money and commitment in order to get one genome sequence. In addition to what we’ve done here, it’s really the work of a lot of other people.