





















Rediscovering Biology: Molecular to Global Perspectives
Online Textbook and Video
This online textbook chapter supports and extends the content of the Human Evolution video. The chapter explores the incredibly small variations in human genetics and how they tell us about our past. Click on the Go button to begin reading, or skip to a sub-section of the chapter in the list below.
1. Introduction
“…we come from a long line of failures. We are apes, a group that almost went extinct fifteen million years ago in competition with the better-designed monkeys. We are primates, a group that almost went extinct forty-five million years ago in competition with the better-designed rodents. We are chordates, a phylum that survived in the Cambrian era 500 million years ago by the skin of its teeth in competition with the brilliantly successful arthropods. Our ecological success came against humbling odds.”
– M. Ridley
We humans have always held a special fascination with our place in the evolutionary pageantry. Where did we come from? That we share close common ancestry with the apes is not in doubt. But what kind of an ape are we? How long ago did our lineage separate from the other apes? Are we still evolving? What can molecular genetics tell us about our history and our future?

Figure 1a. The chimpanzee, humans’ closest living relative.
Courtesy Roger Birkell.
Concerning our place among the apes, Thomas Huxley (known as “Darwin’s bulldog” because of his popularization of Darwin’s theory of evolution) provided an early correct answer in 1863. Huxley placed humans with chimpanzees and gorillas (the great apes of Africa), and separate from orangutans and gibbons. Numerous lines of evidence have since strongly supported this view. Morphologically, we share many derived traits with the African apes, including enlarged brow ridges, elongated skulls, shortened canine teeth, and enlarged mammary glands. We are also much more similar at the DNA level to the African apes than we are to any other species.
Scientists have identified three species of African apes: the gorilla (Gorilla gorilla) and two species of chimpanzees. Pan troglodytes, the common chimpanzee, is the larger of the two. Pan paniscus, which had previously been called the “pygmy chimpanzee,” is now named the “bonobo.” In addition to size, the two chimpanzees differ in social structure and temperament; the bonobos appear to be more peaceful and egalitarian than the common chimpanzees. Both species of chimpanzees use tools; however, the types of tools vary in different populations of both species.

Figure 1b. The gorilla, another living relative to humans.
Courtesy Roger Birkell.
By about 1980 a combination of crude molecular techniques — based on the divergence of certain proteins and the fossil record — allowed us to determine that humans, chimps, and gorillas last shared a common ancestor approximately five million to eight million years ago. Those methods, however, were not able to ascertain the order in which the three species split. Several questions remained. For instance, are chimpanzees and gorillas each other’s closest relatives? Or is the closest relationship between humans and chimpanzees? Or is it between humans and gorillas?
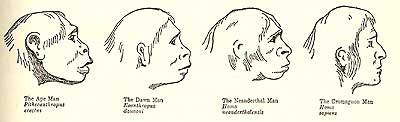
We could encapsulate the outdated view of evolution as progress up a ladder of changes. First the ability to walk upright (bipedalism) appeared. Soon after, the lineage leading to humans (the hominids) split off from the other African apes. Many fossils of the genus Australopithecus demonstrate that the earliest bipedal hominids did not substantially differ from chimpanzees in brain size. In the outdated view of human evolution, there was a slow steady increase in brain size as Australopithecus afarensis (better known as “Lucy”) evolved into Homo habilis, and then into Homo erectus. Brain size continued to increase until the appearance of Homo neanderthalis (the Neanderthals), who looked much like us but had larger brow ridges.
This older view of human evolution is not so much incorrect as it is incomplete and misleading. New fossil evidence demonstrates that the hominid lineage, our family tree, is more bush-like than ladder-like. Studies of these fossils show that several species of hominids co-existed for long periods of time. New molecular genetic evidence allows us to address which two of the three species — chimpanzee, gorilla, and human — represents the two closest relatives. Genetic data also can address the patterns of variation within and among human population. In addition, the molecular genetic data demonstrate how infectious disease has shaped genetic variation in humans.
2. New Fossils
During the 1990s archaeologists unearthed dozens of new fossil hominids. These have been particularly useful for illuminating the changes that took place as the human lineage split from the chimp lineage. One important find was in Ethiopia by Tim White (University of California-Berkeley), Gen Suwa (University of Tokyo), and others who found a fossil, which they determined to be 4.4 million years old. The fossil, Ardipithecus ramidus, probably represents a transitional form with respect to the evolution of bipedalism: while it may have been able to walk upright, it had a different posture than we do. It probably spent some time upright and some time walking like a chimp, on its knuckles. In other respects, it looked much like chimp, except for subtle differences in teeth and skull. The first, clearly bipedal hominids — Australopithecus anamensis and Australopithecus afarensis — appeared about 4.1 million years ago, shortly after A. ramidus.

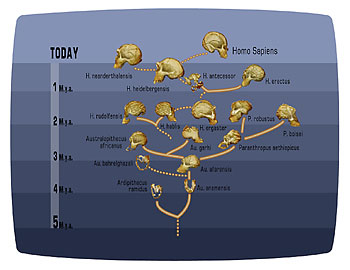
Figure 2. The human “bush,” as postulated from fossil finds of hominid species.
Other fossil discoveries illustrate the bushiness of the human lineage. As seen in the illustration, as many as four different apparent species often lived at the same time (Fig. 2). While there was a general trend toward increased brain size with time, species with considerably different brain sizes lived simultaneously. Questions remain about how different species replaced previous ones. Was it through warfare? Was it that the replacing species were better competitors? Perhaps it was simply a random event. We don’t really know.
Despite the inferences we can draw from these new fossil findings, the fossil record still has limitations; it is incomplete. How can one determine whether different fossils belong to the same species? Species determinations are based on the ability, or the perceived ability, of different groups to interbreed. In cases where it is infeasible or immoral to do experiments crossing the two groups, one can infer the capacity for the groups to interbreed based on genetic data. Yet, with few exceptions, scientists cannot extract DNA evidence from fossils; only morphological characters are available. How then can one make the inferences about the capacity to interbreed? For instance, sexual dimorphism may lead one to classify males and females of the same population as separate species.
3. What Does DNA Tell Us About Our Position Among the Apes?
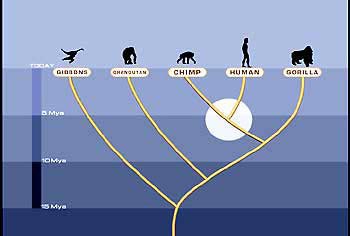
The new genetic data have substantially contributed to our understanding of the relationship between our species and its closest relatives. Based on several independent lines of evidence,

Figure 3. A tree showing the evolution of the hominoids, including the great apes and humans.
we can now say with confidence that humans are more related to chimpanzees than to gorillas (Fig. 3). While the two species of chimpanzees are each other’s closest relatives, their next closest relative is H. sapiens and not G. gorilla.
How do we know this? Evolutionary geneticists have been increasingly able to draw better and more robust inferences about the relationships among different organisms based on morphological and molecular genetic data, and new systematic methodology. These methods have also been used to determine our relationship among the apes. (See the Evolution and Phylogenetics unit.) In essence, groups of organisms (known as taxa) are placed into clades that are nested in larger clades based on shared ancestry. All of the taxa in a given clade are assumed to have a single common ancestor.
The first DNA-based data used to determine the relationships of the African apes came from mitochondria. These intracellular organelles enable animals to use aerobic respiration and have DNA that evolves relatively quickly in mammals. Consequently, mitochondrial DNA (mtDNA) is useful in analyzing the relationships of closely related species and populations within species. Mitochondria are also abundant in cells and, thus, mtDNA was easier to obtain than nuclear DNA.
New DNA amplification technologies developed during the 1990s, such as the polymerase chain reaction (PCR), made obtaining sufficient quantities of DNA much easier. (See the Genetically Modified Organisms unit.) Yet, for historical reasons, most taxonomic studies that used DNA characters were first done with mtDNA. Based on the evidence from mtDNA sequences, chimpanzees and humans were determined to be each other’s closest relatives. These studies further suggest that humans and chimpanzees separated almost five million years ago, and the human-chimp clade separated from gorillas almost eight million years ago.
Critics raised an important point about the inferences based on the mtDNA studies: it is based on only a single, independently evolving gene region. When one considers very closely related groups of species, the constructed phylogenetic tree based on data from one gene may be different than one constructed from a different gene. Either one or both gene trees may not accurately reflect the true evolutionary history of the species. This phenomenon occurs because of genetic variation (polymorphism) in the ancestral species. Ancestral polymorphism can segregate differently in the different descendant species; that is, in one of the different descendant species, one of the variants may become fixed and in another descendant species a different variant may be fixed. Either natural selection or random genetic drift can cause this phenomenon. In either case, there is a possibility that the history of the gene region may not reflect the history of the species. In other words, suppose that chimps really did split first from the lineage containing humans and chimps. It would still be possible that the phylogenetic tree based on a single gene may have gorillas splitting off from humans and chimps, or humans splitting from chimps and gorillas.
In the case of determining the relationships among the African apes, the solution to this challenge was simply the collection of more data from more genes. Mary-Ellen Ruvolo analyzed data sets from fourteen independent gene regions. In eleven of the cases, humans and chimps are each other’s closest relatives (sister taxa). In two cases, gorillas and chimpanzees are sister taxa, and in another, humans and gorillas are sister taxa. Statistical tests show that these results are highly unlikely to arise unless humans and chimpanzees are indeed each other’s closest relatives. Subsequent analyses with even more genes have corroborated the conclusion reached by Ruvolo and the earlier mitochondrial DNA studies: we are closest to chimps.
4. Variation Within and Among Human Populations
At the DNA level, humans are both very similar to and very different from one another. On average, pairs of individual humans share 99.9 percent DNA sequence identity. Due to the sheer size of our genomes, however, we possess numerous differences from one another. The human genome consists of just over three billion nucleotides; that 0.1 percent of difference represents three million variants between the average pair. The vast majority of these variants have no functional significance. However, even if one in a thousand did, that would still mean that we would each differ at thousands of functionally important sites.
How does this variation compare with that of other species? Humans actually have less genetic variation than do their closest relatives. For instance, the average difference between two randomly selected chimpanzees is roughly four times greater than between two humans. This is, at first glance, surprising. Based on population genetic theory, levels of genetic variation within species should correlate positively with population size. This predicted correlation comes about because the strength of random genetic drift — which results in the loss of genetic variation — increases at lower population sizes. Yet, the human population numbers in the billions and the population sizes of chimpanzees and gorillas are fewer than a hundred thousand.
What could explain that discrepancy? The strength of genetic drift is dependent not on the current census population size but on the historical population sizes. The relatively low levels of genetic variation in humans can be explained by a severe, but short-lasting, population bottleneck, where the population of our species was likely reduced to a few thousand. It could also be explained by a more moderate, sustained bottleneck. During this bottleneck, the population was possibly in the tens to hundreds of thousands for a more considerable time. Alternately, natural selection could also either increase or decrease the extent of variation in one of the species. Yet, because it is unlikely that natural selection would act in the same way on multiple regions of the genome, the difference in the extent of genetic variation between humans and chimpanzees is more likely a consequence of historical demography.
How is this variation partitioned according to known racial groups? During the 1970s the then state-of-the-art technique of electrophoresis of protein variants showed that around eighty to ninety percent of human genetic variation was within ethnic populations, five to ten percent was among ethnic populations within the major racial groups, and only about five to ten percent was among the major racial groups. In other words, “if everybody on earth became extinct except for the Kikiyu of East Africa, about eighty-five percent of all human variation would still be present in the reconstituted species”. More recent analyses of DNA sequence data strongly confirm the results of earlier protein electrophoresis studies. In both the protein electrophoresis and the DNA sequence studies, the differences between racial groups are generally ones of frequencies and not kind. The situation of “fixed differences” — in which all individuals in one group have variant A and all individuals of another group have variant B — is extremely rare in humans. Instead, groups vary by having different frequencies of genetic variants. There are cases of “private alleles,” however, where genetic variants are found in low to intermediate frequencies in some populations, but are virtually absent from others.
5. Out of Africa?
As with determining the relationships of the apes, the first DNA-based studies of the relationships of human populations also used mitochondrial DNA. In mammals, mitochondria have an interesting inheritance pattern: they are transmitted nearly exclusively along maternal lines. Although males have mitochondria, they do not transmit them to their offspring.

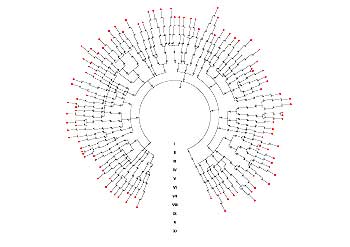
Figure 4. The diagram illustrates how one line of mitochondrial DNA came to be carried by all living humans, passed down to us through the “Mitochondrial Eve.”
Thus, all of your mitochondria came from your maternal grandmother and, by extension, your maternal-maternal great-grandmother. In 1987 Rebecca Cann, Mark Stoneking, and Alan Wilson (then at University of California-Berkeley) published a controversial and provocative paper in Nature, stating that they had located the common ancestor of all mitochondrial variants – the so-called Mitochondrial Eve (Fig. 4). They placed her in Africa approximately 200,000 years ago; subsequent studies have found similar results. Fifteen years after that first paper, the results remain a source of interest and controversy.
Why are the Mitochondrial Eve studies a continual source of controversy within the human evolutionary genetics community? That there is a common ancestor of mitochondrial DNA sequences is not a surprise. In fact, it is a consequence of Mendelian genetics: genes taken from any sample within a population will share a common ancestor. Take pairs of gene copies in the same population. Some of them will share the same ancestor from one generation ago; they are from siblings of the same parents. Some pairs of gene copies will trace a common ancestor two generations back. Some pairs will share common ancestry even further back. However, eventually, all copies will share a common ancestor. What is of interest is how long it takes all gene copies to coalesce to that ancestor.
What the debate focuses on is the timing and the location of the Mitochondrial Eve. The initial studies showed that there was one clade consisting only of African individuals, and one with African and other individuals. Hence, we can infer that the common ancestor lived in Africa. Numerous researchers challenged the methodology of the original study. For instance, the original study used African-American individuals instead of individuals from Africa. Most of the subsequent studies, using more data — including data from individuals from several African tribes – and better methodology seem to confirm that Africa is the location of the common ancestor.
To determine the age of the Mitochondrial Eve, biologists need to first make assumptions about the way evolution proceeds. The usual assumption is that changes in the DNA occur roughly in a clock-like fashion – that there is a so-called molecular clock. The molecular clock assumes that groups separated by twenty nucleotide changes have common ancestors that are roughly twice as old those separated by ten nucleotide changes. No one believes DNA evolution proceeds in a perfect clock-like manner. What is debated is the extent to which the clock assumption can provide an estimate about divergence times. The usefulness and the accuracy of molecular clocks have been controversial ever since Zuckerkandel and Pauling proposed them in the 1960s. Yet, most evolutionary biologists agree that the molecular clock concept has at least some validity.
Inferring dates based on a molecular clock also requires that one calibrate the clock. How quickly do the changes occur in the lineage(s) of interest? Different molecular clocks based on different regions of the genome or different types of organisms don’t all tick at the same rate. To calibrate a molecular clock, researchers usually use a lineage split for which they have at least some degree of confidence about when it occurred. They then divide the amount of genetic divergence by the time when the groups last shared a common ancestor. In the case of mtDNA, the calibration was set by the human-chimp split of six million years. Because chimps and humans differ by about twelve percent of nucleotides in mtDNA, the rate of change for the hominid lineage for mtDNA is about two percent per million years. The average total divergence from contemporary sequences to the inferred sequence of the mtEve is about 0.4% and, thus, the divergence time is about 200,000 years. The confidence limits, however, of this estimate are rather large. Christopher Wills once concluded that it is possible that the upper-end of mtEve’s age may be as much as 800,000 years; new data places his latest estimate at 400,000 years. 3
Different genes will often have different evolutionary histories. One should not expect the male equivalent (“the Y chromosome Adam”) to have lived at the same place and the same time as the Mitochondrial Eve. Owing in part to having a lower mutation rate, the human Y chromosome generally has less variation than the mitochondria, which makes analysis more difficult. Nonetheless, recent studies suggest that the last common ancestor of all existing human Y chromosomes also lived in Africa — but more recently than Mitochondrial Eve.
Largely from the Mitochondrial Eve studies, one model – the out of Africa hypothesis — gained favor among anthropologists and human evolutionary geneticists. This

Figure 5. Top: The “Out of Africa,” or “Replacement,” hypothesis suggests all living humans evolved from a group that originated in Africa. Bottom: The “Multiregional” hypothesis suggests several groups evolved in parallel to form today’s population of humans.
hypothesis, which is sometimes called the “replacement hypothesis,” postulates that modern Homo sapiens spread out of Africa, into Europe and Asia, and replaced archaic Homo sapiens living in those regions (Fig. 5). In contrast, Milford Wolpoff and others have proposed the multiregional hypothesis. They argue that the archaic Homo sapiens populations in the different regions (Europe, Asia, and Africa) all evolved together into modern Homo sapiens. While genetic changes would first occur in one locality, gene flow would spread those changes into the other localities.
The out of Africa and multiregional hypotheses make several distinct predictions. One would predict that under the out of Africa hypothesis, Africa would be the origin of the common ancestor of variants for most of the independent data sets (different genes) tested. The multiregional hypothesis would predict a random pattern. Under the out of Africa model, the divergence time between the African and the non-African populations would have an upper limit of about 200,000 years. In contrast, the multiregional hypothesis would predict a divergence time of approximately one million years. One caveat is that the apparent age of the divergence could be reduced by the gene flow among the populations. Another caveat is that selection can also alter the apparent divergence times. The out of Africa hypothesis also predicts that there will be more genetic diversity within the African population than within the other populations.
As of 2003 the evidence seems to favor the out of Africa model though some intermediate positions cannot be ruled out. In nearly all of the studies more genetic diversity is seen in the African populations than in others. In addition, the divergence times appear more consistent with the out of Africa than the multiregional hypothesis. As we obtain more and more sequences from different regions from the genome, this debate should become resolved.
6. Neaderthals in Our Gene Pool?

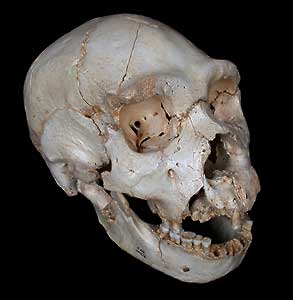
Figure 6a. An artist’s rendering of a Neanderthal man
Zdenik Burian, NEANDERTHAL (1960). Courtesy of Zdeněk Burian, www.zdenekburian.com
Have Neanderthals contributed to our gene pool? This question is related to, but is distinct from, the “out-of-Africa” debate. If Neanderthals had made a substantial contribution to the gene pool of contemporary humans, replacement models like out of Africa would be severely challenged. On the other hand, while the lack of Neanderthal contribution to the contemporary human gene pool would be consistent with the out of Africa model, that particular result alone would not disprove the multiregional hypothesis. It is also possible that there was a substantial exchange of genes across many different human populations but that the Neanderthal population was not involved.
How can we tell whether Neanderthals contributed to the contemporary gene pool? You can’t get DNA from fossil humans.

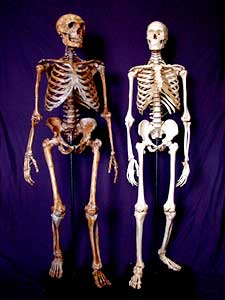
Figure 6b. A Neanderthal and an adult human skeleton side-by-side.
Or can you? Data from fragments of DNA collected from different Neanderthal fossils have led to the conclusion that Neanderthals probably did not contribute to the contemporary gene pool. In 2000 Igor Ovchinnikov and his colleagues were able to obtain small fragments of mtDNA from a 29,000-year-old Neanderthal fossil found in the Caucasus Mountains. They compared the mitochondrial sequences from their fossil to mtDNA collected from a previously collected Neanderthal fossil from Germany. Ovchinnikov and his colleagues concluded, “Phylogenetic analysis places the two Neanderthals from the Caucasus and western Germany together in a clade that is distinct from modern humans, suggesting that their mtDNA types have not contributed to the modern human mtDNA pool.”
7. Human Genetic Variation and Disease
Disease has continued to have a strong impact on human mortality and reproduction. One would expect there to be genetic variation for the ability to resist disease. Indeed, there is such variation. Moreover, biologists have been increasingly able to correlate variation at specific genetic loci, and susceptibility to or severity of various diseases. For example, scientists are combining genetic and genealogical data to locate genes that affect disease tendency in Icelanders.
Genetic variation for disease resistance and natural selection associated with disease has shaped the evolution of our species. Below, we discuss the impact of two infectious diseases: malaria and HIV. We conclude with a discussion of the genetics of asthma propensity — an illustration of the complex interplay of genetics and environmental effects.
8. Malaria, Sickle Cell Anemia, and Balancing Selection
Sickle cell anemia affects approximately 70,000 Americans, almost exclusively those with African ancestry. The lifespan of an individual with sickle cell anemia is currently approximately 40 years in the United States. Before the advent of modern medicine, individuals with the disease usually died before they could have offspring.
The disease iscaused by a change in a single amino acid difference in the beta chain of hemoglobin. Individuals with two copies of the sickle form of the gene have sickle cell anemia. Heterozygotes — individuals with one normal and one mutant copy of the gene — appear normal and do not manifest the disease except under very stressful conditions; however, they are carriers. If two carriers have a child, the child has a twenty-five percent probability of receiving two copies of the sickle form and having the anemia. Approximately ten percent of African Americans are carriers. In Africa itself, the frequencies of the disease and carriers are even higher.
If sickle cell anemia is so deadly, why are so many people heterozygous carriers of the disease? Moreover, why does the disease afflict predominantly one racial group? Surpisingly, the answer has to do with malaria. Heterozygote sickle cell carriers are much more resistant to malaria than those with just normal hemoglobin. Because heterozygotes have the best of both worlds (no sickle cell anemia and higher malaria resistance) and malaria is extremely prevalent in Africa, the sickle allele can be maintained in balance with the normal allele. Note that in the United States, where malaria is rare, the carriers possess no such advantage and may even have a small selective disadvantage. Therefore, due to the strong selection acting against those with the anemia, the frequency of sickle cell anemia should slowly decline in the United States. That the frequency of the sickle cell allele is higher in African populations than in African Americans is due to both this selection and the genetic mixing between whites and blacks in the United States.
This situation, where selection actively maintains two or more alleles at a locus, is called balancing selection. Balancing selection can arise by the heterozygotes having a selective advantage, as in the case of sickle cell anemia. It can also arise in cases where rare alleles have a selective advantage. In extreme cases, balancing selection can maintain alleles in populations long enough for speciation to occur. In such cases, one species may have alleles that are more similar to those of the other species than they are to other alleles

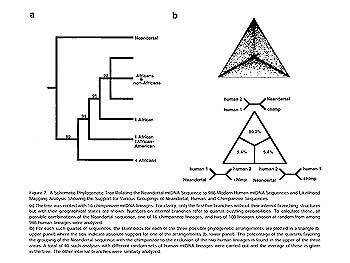
Figure 7. For nearly all genes, human alleles cluster together and chimp alleles cluster together (left). In the case of the major histocompatibility complex (MHC), human alleles are often more closely related to chimp alleles and vice-versa. This occurs due to balancing selection maintaining variation at the MHC (right).
of the same species. One case of this phenomenon occurs at loci at the major histocompatibility complex (MHC) wherein some human alleles are much more closely related to some chimpanzee alleles than they are to other human alleles (Fig. 7). MHC – also called the human leukocyte antigen (HLA) loci when referring to it in humans — encodes proteins that are used to recognize foreign invaders by cells of the immune system. Chimp-like alleles have been maintained in the human population not because they are chimp-like, but because either having rare alleles or having two different alleles has provided a selective advantage. This balancing selection is so powerful that alleles are maintained that predate the human/chimp split.
9. Resistance to HIV
Despite the lethality of HIV/AIDS, susceptibility to HIV infection and progression to AIDS is rather variable. There are individuals who have been exposed to HIV multiple times but who either remain uninfected or if they are infected, progress more slowly to full-blown AIDS. Recent studies have shown that some of the variation in HIV resistance has a genetic component.
HIV operates by subverting the immune system; therefore, it is logical that differences in the immune system may play a role in the genetic variation of resistance to HIV. Indeed, some HIV-resistant individuals possess different chemokine receptors than HIV-susceptible individuals. What’s a chemokine receptor? First, let’s discuss chemokines.
Chemokines are molecular signals released by cells of the immune system that stimulate white blood cells to move to inflamed tissues. They are metaphoric “cries for help.” The chemokines bind to receptors located on the white blood cells. Macrophages – those white blood cells that engulf foreign particles and are an early stage of defense — possess the chemokine receptor that is encoded by the gene CCR5. By subverting the normal function of this chemokine receptor, HIV is able to gain entry into macrophages. (See the HIV and AIDS unit.)
Individuals that have lower expressions of this protein due to variants of the CCR5 gene have an increased resistance to HIV; their macrophages are metaphorically more cautious about the signals they respond to. The most obvious case of a “more cautious” CCR5 variant is the allele that has a deletion of thirty-two nucleotides. Individuals who are heterozygous for this variant, CCR5-delta32, have substantially increased resistance to HIV infection; if infected, progress to full-blown AIDS is much slower than normal. Individuals that are homozygous for CCR5-delta32 are virtually completely resistant to HIV. In European populations about twenty percent of individuals are heterozygotes, and one percent are homozygotes in some populations. In contrast, the allele is rare in the Asian populations and virtually absent in the African populations.
Why is this deletion variant present in some populations in such high frequencies? HIV is, at most, a couple of centuries old and, more likely, less than a hundred years old. That isn’t sufficient time for natural selection to increase the frequency of a rare allele, such as is observed in the European populations. Furthermore, the selection pressures caused by HIV should be much higher in Africa than in Europe. It is also probable that the decreased receptivity to chemokines would be somewhat costly. Some biologists have suggested that the deletion allele could be a vestige of plague resistance. It may have led to increased survival during the Black Plague of the fourteenth century in Europe, and has had an unintended — but welcome — consequence of HIV resistance. The increased frequency of the variant in Europe would be consistent with that scenario.
The environment, and in particular, disease has continued to exert strong pressures on human populations. Generally, we are unable to directly observe changes in species because these changes occur in time scales that exceed human lifespans. Yet, we may be able to detect small changes in allele frequencies that have occurred in populations due to epidemics.
10. The Genetics of Asthma, a Complex Disease
Asthma, which can be considered a consequence of an overly sensitive immune system, is a substantial and growing health problem. As of the year 2000 it was the eighth-most prevalent chronic disease in the United States and affected about fifteen million Americans. That’s an increase of more than fifty percent between 1982 and 1996. While this dramatic increase underscores a clear environmental component to asthma, asthma is also a genetic disease. The likelihood for getting asthma varies widely and has been known to run in families. Identical (monozygous) twins have a higher concordance of their asthma susceptibility than do fraternal (dizygous) twins.
The genetics of asthma, like the genetics of most prevalent diseases, is complex. There is no single gene for asthma, coronary heart disease, or most forms of cancer. Moreover, the severity of asthma-related symptoms follows a continuum. During the 1990s geneticists have been increasingly able to map complex, continuous conditions to regions of the genome. About a dozen different regions of the genome have been identified for having effects on asthma susceptibility. Interestingly, asthma propensity maps to different genetic regions depending on which ethnic group(s) are studied. As summarized by Matt Ridley, “the gene that most defined susceptibility to asthma in blacks was not the same that most defined susceptibility in whites, which was different again from the gene that most defined susceptibility in Hispanics.” (p. 75)
Why could this be? Michael Wade presents a plausible explanation for this failure to replicate the results in different populations: that the genetic background is different across the different populations. 5 The different populations could have somewhat different allele frequencies of genes that act as modifiers of the genes that have a large effect on asthma propensity. This would be consistent with we know about genetic variation in human populations: differences among populations are usually ones of frequency, not of kind. Because of different frequencies of modifier alleles across the populations, a particular gene may explain more of the variation in asthma sensitivity. Determining whether this is the explanation for the different results obtained for asthma susceptibility will require first isolating the modifier genes and then testing whether their frequencies vary in different populations.
11. Our History, Our Future
The common ancestor that we shared with chimpanzees about six million years ago was much more like modern chimps than us. In our lineage, the hominids, so many changes occurred: bipedalism, substantially larger brains, tool use, language, and so on. The genetic bases of these important transitional changes remain murky at best. What genetics has shown us is that we are one species, somewhat lacking in genetic variation, and having only slight differences among different populations. Genetic studies have also shown that disease and other factors continue to substantially affect our evolutionary trajectory.
End Notes
-
- Ridley, M. Genome: The autobiography of a species in 23 chapters. Perennial.
- Lewontin, R. C., S. Rose, and L. J. Kamin. 1984. Not in our genes: Biology, ideology and human nature.
- Wills, C. 1995. When did Eve live? An evolutionary detective story. Evolution 49:593-607.
- Ovchinnikov, I. V., A. Gotherstrom, G. P. Romanova, V. M. Kharitonov, K. Liden, and W. Goodwin. 2000. Molecular analysis of Neanderthal DNA from the northern Caucasus. Nature 404:490-93.
- Wade, M. J. 2001. Epistasis, complex traits and mapping genes. Genetica 112/113:59-69.