


Rediscovering Biology: Molecular to Global Perspectives
Cell Biology and Cancer Expert Interview Transcript: Leland Hartwell, Ph.D.
 Director, Fred Hutchinson Cancer Research Center
Director, Fred Hutchinson Cancer Research Center
Dr. Hartwell is presently the president and director of the Fred Hutchinson Cancer Research Center in Seattle. He won the Nobel Prize in Medicine in 2001 for his work on control of the cell cycle. Using the yeast as a model organism, he has identified many genes responsible for controlling cell division.
Interview Transcript
Interview with Leland Hartwell, Ph.D. Dr. Hartwell is presently the president and director of the Fred Hutchinson Cancer Research Center in Seattle. He won the Nobel Prize in Medicine in 2001 for his work on control of the cell cycle. Using the yeast as a model organism, he has identified many genes responsible for controlling cell division.
In 1970 the government declared the war on cancer; Can you tell me a little bit from that year to today, what have we learned?
There [are] some people [who] have expressed disappointment that we didn’t cure cancer the way we put a man on the moon when Kennedy declared we would do that within the decade. The problem was that putting a man on the moon was an engineering problem and the science was completely understood.
The difference with cancer is that it really was a scientific problem in 1970. And we didn’t know how to solve it, how to understand it. The answers to scientific problems can’t be predicted. For example, unifying all the physical forces that physicists work on, no physicist will tell you when that problem will be solved. What we’ve learned since then I think is really profound.
We now understand a lot about cancer. We know that cancer arises from a single cell out of the 1013 cells we have in our bodies. We know that it results from a series of genetic changes — at least a half a dozen or so accumulate over our lifetime. We know that those genetic changes alter half a dozen different cellular processes and we know what those processes are-[processes] having to do with cell division and growth control and genetic instability, mortality, the suicide mechanism in cells; the ability of the cells to migrate; the ability of the cells to attract to them a blood supply. And so that’s pretty profound that in a few sentences one can summarize a sophisticated, fundamental understanding of what a cancer is. So I’d say we made great progress and that some aspects of cancer research are really becoming more like engineering problems because we have some fundamental understanding and we now can sort of map a path to what we need to do to treat the disease better.
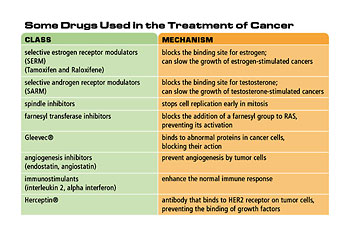
How has our acquired knowledge of how cancer works enabled us to develop new cancer drugs like Gleevec?
Well, the sort of standard paradigm for how we’re going to cure cancer is small molecule therapy. The idea is that you find out what is different about a cancer cell in terms of its genes and its proteins and you build a small molecule that inhibits an essential difference in the cancer cell. Then you inhibit the cancer and that’s it. That is the model of Gleevec and it’s the only case we can point to where it’s worked.
It’s the current poster child and the question is whether it’s going to be a general paradigm or not. But the chronic myelogenous leukemia that Gleevec is very effective against is a genetic translocation that hooks up to parts of a particular gene. [This] new gene is expressed in the cancer cell is not present in any of the normal cells. So it is an absolutely unique target. Gleevec is an inhibitor of that particular enzyme. I think there are a lot of other ways to effect [an] advantage for cancer patients other than this sort of standard, small molecule therapy.
What would you say, in your field of cancer research, is the big question that researchers are all trying to answer?
I don’t think there is a single, big question. I think if I had to reduce it to a single big question I would say it is understanding the complexity of biology. But there is a lot hidden in that word complexity.
Can you describe how we came to understand that cancers are multi-hit genetic diseases?
One of the fundamental aspects of cancer that’s been known for a very long time is that the risk of cancer increases dramatically with age. Why is that? Our current understanding is that it’s because a cell has to accumulate a whole series of changes before it actually becomes a malignant cancer cell. And we understand what many of these changes are in terms of cell physiology. If it were the case that you could change one gene or one aspect of a cell and get cancer, we would [get] cancer before we were visible as a human being in early development. We have so many cells in our body and there are inaccuracies in cellular reproduction. If a single gene mutation could convert [a cell] to a cancer we would get cancer, very, very early. So over the course of evolution safeguards must have built up in multi-cellular organisms to allow them to have large numbers of cells. And the simple answer has been that no one change will convert a human cell to a cancer cell. It takes many changes and that is the safeguard that is necessary so that normally even with the huge number of cells that we have, cancer only occurs relatively late in age.
Can you talk about where we get these mutations from?
Well the reproduction of our DNA is like reproducing an encyclopedia. When scribes used to sit and copy old books, they made mistakes. And those mistakes got propagated as a result of copying errors. So there is always a mistake rate in any cellular process. There is a mistake rate in the cells replicating its DNA. The mistake rate is extremely low-it’s estimated to be about 1 mistake in copying [for every]1010 basis. That is an intrinsic error rate. It is estimated to be a mistake every cell division, somewhere in our DNA. There is so much DNA to copy. The systems that have been put in place to make that process accurate are multitudeness and the rate of mistakes of just copying DNA in a test tube is tens of thousands of times greater than that.
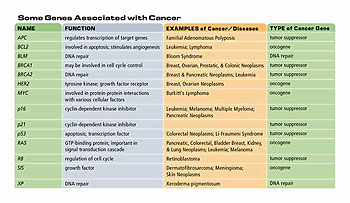
We have figured out the role of several oncogenes and tumor suppressor genes in cancer. What are we trying to figure out now?
Well, I would characterize our current state of knowledge as one where we have a broad outline that is probably right. That cancer results from half a dozen, genetic changes to certain cellular processes that we understand to some extent. What is missing is a real depth of knowledge. So we don’t know all the genes that are involved in any one of these cellular processes and we don’t know all the possible mutations that can occur to inactivate each of these cellular processes.
We have a great deal to learn about the details and it’s sort of like taking a week vacation to Sydney and say you’ve been to Australia. You know if you really want to understand Australia you’re really going to have to spend more time than that. We are sort of at the one-week vacation level I think in terms of the complexity of cells.
Stepping away from this field’s individual research problems, are there moments in which you are filled with awe and wonder at what has been figured out?
I’m always filled with awe about what we haven’t figured out than what we have figured out. There are [the] cellular processes-you learn from disease how accurate the “normal” is, which you would only appreciate by the fact that it goes awry occasionally and produces disease. The tremendous mystery that I think will always exist [is the layers] of complexity which are just unbelievably enormous. A cell has tens of thousands of proteins, each performing a different function. Each of those proteins interact with 10 other proteins to control one another. The matrix of interactions is incredibly complex.
But on top of that, every individual in the population is genetically different. So you have complexity layered on complexity, layered on complexity. And I think we’re very arrogant in the sense that we think we understand a lot. But if we [weren’t] arrogant, we wouldn’t have the courage to proceed and try to do the next experiment to understand the next thing if we really were overcome by the enormity of the task.
What did we used to think about the way the cell cycle proceded and what do we know now thanks to your work and the work of others?
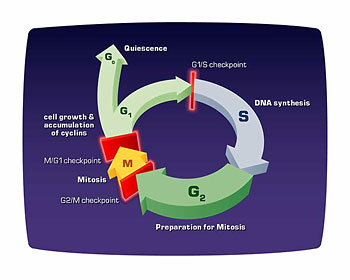
Cell division was originally appreciated primarily from a microscopic observation. You could see things happening and you could see primarily the chromosomes of the cell that contain the DNA, condensing at one time in the cell cycle and segregating. You knew by the time the cell would reach its next division, that all those chromosomes had to be duplicated again. So the focus was really on what you could see. The molecular insights into that have primarily come out of understanding the cycle and independent kinase, which is sort of the clock of the cell cycle.
And a dependent protein degradation system that removes proteins at certain times. We now have a molecular appreciation of how these events are orchestrated. But with respect to complexity, I think this is just the tip of the iceberg. What we don’t understand at all is the fact that everything in the cell is duplicated, not just the chromosomes. Every cell always comes from a pre-existing cell and traces its history back three billion years. So what is it that one cell contributes to the next cell, besides the DNA?
What about the cell cycle clock?
The basic orchestration of the cell cycle really requires solving two kinds of fundamental problems. One, is that you have to establish order so that certain things occur before other things. DNA replicates before it segregates. The second thing you have to do is coordinate the activities of very large numbers of proteins. DNA replication involves thousands of proteins probably. And chromosome segregation probably involves a thousand proteins.
So how do you get all of those to function together at the right time?
Partial answers to those two questions have to do with the cycle independent kinase. A kinase that can phosphorolate many, many proteins is activated at a certain time in the cell cycle. I think the major insights into this story come from Paul Mercer’s laboratory. A kinase can phosphorylate many, many different proteins. That solves the problem of how you orchestrate thousands of different proteins to function at a certain time.
Then the question of order comes from the fact that the kinase’s activity is dependent upon the kinase interacting with a cyclin, which says which proteins it can phosphorylate. [When the cell] activates DNA synthesis, the kinase combines with a cyclin that has specificity for the proteins that are involved in DNA replication. Then once DNA replication takes place, that cyclin is destroyed so you can’t replicate DNA again. And a new cyclin comes on that will activate the phosphorylation of the proteins needed to segregate the chromosomes.
So that explains why things are ordered. It’s a very beautiful story and undoubtedly too simple, but you can achieve some rather fundamental insight into the problem of coordinating many different things and then ordering different events with a very simple mechanism.
Can you explain more about checkpoints. What are they and why are they important?
I’ve always been motivated in my research by aspects of cancer and wanting to understand the fundamental mechanisms. At first, it seemed obvious that cell division [in cancer] was a basic problem, and I wanted to understand something about cell division. Later I became fascinated with the concept of genetic instability in cancer. This is really a fundamental aspect of cancer. Since we know now that a half a dozen or so changes have to take place in the genome, we also know that those changes would never occur in the history of a single cell over our lifetimes given the normal accuracy of cell division. The reason that they can occur in a cell is because one of the changes that takes place is genetic instability.
It makes the cell more unstable so it creates more changes than a normal cell would. This is very characteristic of any system that has to evolve quickly.
The AIDS virus and other pathogens all have an intrinsic instability. Even our immune system, which has to adapt to many different insults, has an intrinsic instability to generate that diversity.
So I became interested in this question of why cancer cells are genetically unstable, when normal cells are very stable. That led to research around how cells handle damage and why they don’t transmit the damage that they receive to their daughter cells? That led us to discover that when yeast cells receive damage, they stop dividing and they repair the damage before they divide. So the cell is aware of the fact that its DNA has been damaged. It controls its cell division to wait and repair.
We began investigating that phenomenon and discovered this control mechanism that recognizes damage and then controls division and response to it. There are probably many such checkpoint mechanisms in cells. There is a half a dozen known now. But there are probably many more in biological systems where the whole system is cognizant of what’s taking place in other parts of it and will stop and coordinate everything if something goes awry.
Doesn’t the protein p53 work in this capacity, to signal the cell that it is damaged? And isn’t p53 implicated in many cancers?
P53 is the most common tumor suppressor mutation in cancer. Many cancers are altered in a gene called p53. And p53 defects in cells really allow cells to bypass two of these controls because p53 is a checkpoint protein. It allows the cells to recognize DNA damage, induces responses to it, and controls cell cycle progression while it makes repairs. But in addition to that, p53 is also responsible for inducing cell death when a cell has received too much damage.
By knocking out p53 you are not only knock out the suicide mechanism that protects us from damaged cells, but you also knock out the mechanism which prevents cells from transmitting damage, so you get the propagation of damage in a way that normal cells don’t do. It’s probably that two for one that makes p53 a particularly vulnerable gene for mutation to cancer.
Can you talk about CDC28?
We [began to work] with yeast cells because they were susceptible to genetics; because they would grow as haploid cells. We couldn’t do the same kind of genetics with human cells. We were looking for the things in common between yeast and human cells. One of the important characteristics of human cells for controlling their division is that they arrest in what is called G1 prior to replicating DNA. When they are not dividing-most of the cells in our body are in G1 and not dividing-they may sit there for months or years before they get a signal to divide. Then they divide and they go back to G1 and they wait [again]. We found that budding yeast cells also control their division in G1 in response either to nutrients or in response to a cell that they are going to mate with. Yeast cells come in two mating types and then cells are normally dividing. But if they come in the vicinity of one another, they each emit hormones that arrest the division of the other cell. So the cells are synchronized and they fuse and make a diploid cell. Those hormones also arrest division in G1, so we called this point of control in yeast cells “start,” because it was the point in which control occurred. We were very pleased that it was in parallel with what had been seen in human cells. It turned out that the gene that controlled the “start” event-CDC28-did encode a cyclin-dependent kinase.
Can you discuss more about yeast and how you chose it to be the model organism to learn about the cell cycle?
Yeast is really a delightful organism to work with and maybe characteristic of science’s beauty in that when you get down to the fundamentals in some way, it’s simple. This is what mathematicians always say of equations-that fundamental equations are beautiful.
Yeast is that way; our work with yeast involves very simple tools. Petri dishes that we grow them on and toothpicks that we isolate them with and pieces of velvet that we replicate the colonies from one plate to another. Most of the conclusions are reached by just looking at whether cells grow or not. Did they grow under this condition or not? From this growth, you can build pretty elaborate stories about what is going on in the cell. They also smell good.
Can you provide an overview of angiogenesis?
Angiogenesis is the growth of blood vessels and all of our tissues have to have a blood supply within a few cell diameters to provide nutrients and oxygen. Tumor cells form a ball of cells that eventually gets big enough [to] becomes limited for nutrients in the middle.
The cells in the middle will start dying because they are not getting fed and they are not getting oxygen. Tumors would be limited to a very small size about the size of a BB if they weren’t able to get fed-so by processes which are still somewhat mysterious, tumor cells emit molecules which attract blood vessels to start growing into them and feeding them. Otherwise, they wouldn’t grow larger. So the process of trying to inhibit [angiogenesis] is very prominent [in] for cancer therapy.
So far the attempts to use that as a therapeutic mechanism have not been successful. It’s still very promising and most certainly [will] become a part of our therapy at some time. The big advantage of focusing therapy on the angiogenesis is if the therapeutic is acting against the normal blood vessel cell rather than the genetically unstable tumor cell the blood vessel cell will remain sensitive to the inhibitor you’ve produced. Whereas the cancer cell being genetically unstable will eventually become resistant to the inhibitor. So it’s a very smart approach to target the normal cell rather than the abnormal cell.
Where do you see the biggest hope in treatment for cancer?
I see the biggest hope for rapid progress in reducing mortality from cancer in the area of early detection. If you look at the survival statistics, typically people who are diagnosed at stage one cancer have 90% survival over five to ten years. Whereas people who are diagnosed at stage four have 10% survival roughly. So we have almost a factor of 10 to gain by diagnosing cancer early rather than late. And the question is how to do this. I believe that the real promise comes from looking in body fluids, probably serum, for the markers that cancer cells emit, the proteins that are present in serum [that are] characteristic of cancer. We’re putting a big effort into doing that by taking cases of 100 people who have a certain type of cancer and 100 people who don’t, comparing their serums, and looking for markers that are characteristic of cancer. [We also look] in the serum of people prior to having gotten cancer to find out how early we can see that signature. And this is the area in which modern molecular and genomic technology can most rapidly influence cancer survival.
What other information would you like to impart to the public about cancer?
There are two things that I think we have learned over the last few decades that are worth keeping in mind. One is how much we can learn about ourselves by studying very simple biological systems-by studying yeast cells. We learned about the genes that control cell division. The Noble Prize this year was given for learning about cell death, apoptosis. That was discovered in small worms. The process about ends of chromosomes and cell mortality that was discovered in protozoa.
So the wonderful thing about biology is it’s unity and our relatedness to all living things and the fact that we can learn about ourselves often much easier by studying other living things. The second thing that the public needs to keep in mind is that advocates [play] a very useful role [in that] they advocate for more research for disease; [however], they are often advocating for a very specific disease, like breast cancer or Parkinson’s disease. The fundamental biology which underlies disease and the fundamental technologies that we can use to discover the answers are very broad and it doesn’t help very much to be dividing things up into very targeted research areas.
For example, I mentioned looking at the serum for proteins that are markers of disease. This will be one technology platform that will apply to every disease. So it’s better to keep a broad mind about what we’re studying.
Everybody is waiting for the day they announce the cure for cancer. Is that ever going to happen?
No. Cancer is much more complicated than to say that there is a cure. People estimate 300 different types of cancer. There are some fundamental commonalities to them all and fundamental ways of understanding and studying them all. But still they each have their own specific history and we do have to solve them individually.
Is there anything else that you would like to impart to teachers?
I think it’s important to also be aware of the fact of how much more we have to learn. I think one of the downsides of education is that we tend to talk about what we understand because it’s hard to talk about what we don’t understand. And because we understand a fair amount we can fill up students’ time with a lot of facts and a student can get the impression that we understand everything.
The best way to destroy that impression is to allow students to ask their own questions. So any set of grammar school kids can start asking me questions about the cell cycle or biology and it will not take very long before I will get to the point where I am saying we don’t know the answer to that. That’s what’s really fundamental. There is certainly far more about the cell division process that we don’t know than what we do know.
It takes a special imagination and creativity in a teacher to be able to communicate the fact that the boundaries of our knowledge are really quite close.