


Rediscovering Biology: Molecular to Global Perspectives
Online Textbook and Video
The online textbook chapters support and extend the content of each video. The Web version can be viewed as a full chapter or as individual sub-sections, and includes links to glossary terms and other related material.
1. The Human Genome Project
Unit 1 / Section 1
” …the acquisition of the sequence is only the beginning. The sequence information provides a starting point from which the real research into the thousands of diseases that have a genetic basis can begin.”
– J. Craig Venter
In 1986 Nobel laureate Renato Dulbecco laid down the gauntlet to the scientific community to sequence the complete human genome. “Its significance,” he said, “would be comparable to that of the effort that led to the conquest of space, and it should be carried out with the same spirit.”
Dulbecco also argued that such a project should be “an international undertaking, because the sequence of the human DNA is the reality of the species, and everything that happens in the world depends upon those sequences.”
Like the conquest of space, sequencing the human genome required the development of wholly new technologies. The human genome, containing more than three billion nucleotides, is vast. In 1986 DNA sequencing had yet to be automated and, consequently, was slow and tedious. Moreover, computer software for sequence analysis was just being developed. Similar to the Apollo project that met President Kennedy’s goal of a manned lunar landing by 1970, the genome project also succeeded — beyond the dreams of the scientists who proposed it.
During the 1990s rapid progress was made in developing automated sequencing methods and improving computer hardware and software. By 2003 biologists had sequenced genomes from about one hundred different species. These species included dozens of bacteria and other microbes, as well as the model systems: yeast, fruit fly, nematode, and mouse. The capstone, of course, was the completion of the human genome sequence. In 2001 two rival teams jointly announced the completion of a draft sequence of the entire human genome, consisting of more than three billion nucleotides.
Is human DNA “the reality of the species”? Do we now have all the information we need to define human life? Perhaps surprisingly, the answers are no. Genetics is more than just DNA. While DNA is the blueprint for life, proteins carry out most cellular functions; DNA just codes for RNA, which codes for protein.
One major surprise emerged from the sequencing of the human genome. Although some scientists expected to find at least 100,000 genes coding for proteins, only about 30,000-35,000 of such genes appear to be in the human genome. These genes comprise only about two percent of the entire DNA. What is the rest of the DNA doing? Biologists once thought that this noncoding DNA was just junk, and hence called it “junk DNA.” As we will see below, evidence now suggests that some junk DNA may have functions.
The quest to understand the workings of human cells will not be over until we understand how this genetic blueprint is used to produce a particular set of proteins – the proteome – for each type of cell and how these proteins control the physiology of the cell. (See the Proteins and Proteomics unit.) We should think of the human genome as a database of critical information that serves as a tool for exploring the workings of the cell and, ultimately, understanding how a complex living organism functions.
2. Sequencing a Genome
Unit 1 / Section 2
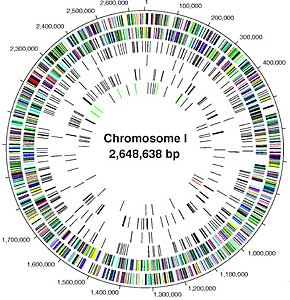
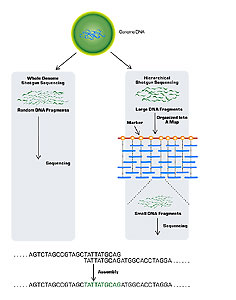
Sequencing a genome is an enormous task. It requires not only finding the nucleotide sequence of small pieces of the genome, but also ordering those small pieces together into the whole genome. A useful analogy is a puzzle, where you must first put together the pieces of a smaller puzzle and then assemble those pieces into a much larger picture. Two general strategies have been used in the sequencing of large genomes: clone-based sequencing and whole genome sequencing (Fig. 1).

Figure 1.
In clone-based sequencing (also known as hierarchical shotgun sequencing) the first step is mapping. One first constructs a map of the chromosomes, marking them at regular intervals of about 100 kilobases (kb). Then, known segments of the marked chromosomes (which can contain very small fragments of DNA) are cloned in plasmids. One special type of plasmid used for genome sequencing is a bacterial artificial chromosome (BAC), which can contain DNA fragments of about 150 kb. The plasmid’s fragments are then further broken into small, random, overlapping fragments of about 0.5 to 1.0 kb. Finally, automated sequencing machines determine the order of each nucleotide of the many small fragments.
Data management and analysis are critical parts of the process, as these sequencing machines generate vast amounts of data. As the data are generated, computer programs align and join the sequences of thousands of small fragments. By repeating this process with the thousands of clones that span each chromosome, researchers can determine the sequences of all the larger clones. Once they know the order of all the larger clones, the researchers can join the clones and determine the sequence of each chromosome.
Finding the sequence of the smaller clone fragments is relatively easy. The challenge is assembling all the pieces. The National Human Genome Research Institute (the public consortium headed by Francis Collins) used clone-based sequencing for the human genome. In doing so, they relied heavily on the work of computer scientists to assemble the final sequence.
Whole genome shotgun sequencing skips the mapping step of clone-based sequencing. Instead, it (1) clones millions of the genome’s small fragments in plasmids, (2) sequences all of these small overlapping fragments, and then (3) uses computers to find matches and join them together.
Celera Genomics, a private company headed by J. Craig Venter, used this approach to clone the human genome. Although they started much later than the public consortium, Celera completed its draft sequence at about the same time as the consortium; however, it had the advantage of having access to all the consortium’s maps.
Genome sequencing projects now generally use some combination of chromosome mapping, and clone-based and whole genome shotgun sequencing of smaller fragments. The technology developed for sequencing the human genome – both in terms of sequencing DNA and in the software and hardware used to assemble the sequences into a genome – has resulted in the rapid sequencing of many other genomes.
3. Finding Genes
Unit 1 / Section 3
Imagine the genome as an encyclopedia with a volume for each chromosome. If you were to open a volume, you would find page after page containing only four letters – A, T, G, and C – without spaces or punctuation. How could you read such a book, or even identify possible words and sentences? The genome sequence itself does not provide direct information on the location of a gene, but there are clues embedded in the sequence that computer programs can find.

Figure 2.
Most simple gene prediction programs use several pieces of sequence information to identify a potential gene in a DNA sequence. The programs look for sequences in the DNA that have the potential to encode a protein. These sequences are called open reading frames (ORFs). An ORF usually begins with a codon of AUG, and then contains a long sequence of codons that specify the protein’s amino acids. The ORF then ends with a stop codon of UAA, UAG, or UGA (Fig. 2). Using overlapping frames of three nucleotides each, the computer program searches the database until it identifies an ORF region. For example, the sequence “abcdefghijk” could be read in three-letter “words” of “abc-def-ghi'” “bcd-efg-hij,” or “cde-fgh-ijk.” Computer programs can scan DNA sequences quickly, using these overlapping reading frames on both the original strand and on the complementary strand, producing a total of six different reading frames for any sequence.

Figure 3. RNA Processing
Using these programs to find ORFs in bacterial genomes is relatively easy. Here, the DNA sequence matches the mRNA. The situation is more complicated for eukaryotic genes, which often contain one or more noncoding regions (introns). To find ORFs in these genes, the introns are removed in a process called splicing (Fig. 3). The final spliced mRNA, which encodes the protein product of the gene, is smaller than the original RNA transcript that matches the genome. The introns are removed, leading to the splicing of the coding regions of a gene (exons) together into the final mRNA. The problem is that a simple ORF-finding program cannot be used with genomic DNA that has introns because those genes do not match the mRNA. While computer programs can identify eukaryotic genes with introns, they are not always accurate.
An alternate approach to characterize genes in eukaryotes is to first make a DNA copy of the mRNA encoded by the gene. To do this, one uses an enzyme called reverse transcriptase. The copy, called cDNA or complementary DNA, has the same sequence as the mRNA, except that the U is replaced by a T. Because the cDNA lacks introns, the sequence of the cloned cDNA can be used to find an ORF. In addition to simply identifying ORFs, many advanced sequence analysis programs use other information to help identify eukaryotic genes in the chromosome. (See the BLAST section below.)
4. Is the Eukaryotic Genome a Vast Junkyard?
Unit 1 / Section 4
Bacteria have small, compact genomes, rich in genes. These genes have fewer noncoding regions and no introns. Eukaryotic genomes, however, often have much more DNA content than prokaryotic genomes. While eukaryotes generally have more genes than bacteria, the difference in gene content is not as great as the difference in DNA content: there is much more noncoding DNA in eukaryotes. In fact, gene-coding regions comprise only about two percent of the human genome.
Most eukaryotic genes are interrupted by large introns. Even with these introns included, however, genes comprise only about twenty-five percent of the human genome. In eukaryotes, repeated sequences characterize great amounts of noncoding DNA. Some of this repetitive DNA is dispersed more or less randomly throughout the genome. There are also millions of copies of other, shorter repeats, but they are typically found in larger blocks. Some trinucleotide (3 bp) repeats are associated with diseases such as fragile X and Huntington’s disease, which result from extra copies of the repeat sequence.
Most of these repeat sequences are transposable elements, that can replicate and insert a copy in a new location in the genome. The result is the amplification of these repetitive elements over time. Transposable elements can be harmful because they can cause mutation when they move into a gene. They also use cellular resources for replication and expression. Are these elements unwelcome guests gone wild or may they actually be useful components of the genome? We don’t really know, but there are some tantalizing suggestions of functions for some of these elements. About one million copies of the repetitive DNA element called Alu repeats lurk in the genomes of each one of us. What are they doing? One study found that these bind to proteins used to reshape chromatin during cell division. Perhaps this apparent junk DNA is actually helping provide structure to the chromosome and regulate the production of proteins in different cell types.
Genomes differ in size, in part because they have different proportions of repetitive DNA. For example, the total genome size of the puffer fish is about one-tenth the size of the human genome. However, the puffer fish genome has about the same number of genes as the human genome, and the genes appear to have the same functions. The puffer fish genome is also smaller than the human genome. This is partially because it contains only about 15 percent repetitive DNA, while more than half the human genome is repetitive DNA. Because most human genes are present in the puffer fish and the puffer fish genome is less cluttered by repetitive DNA, this model organism may help scientists identify the genes responsible for human diseases.
5. The Difference May Lie Not in the Sequence but in the Expression
Unit 1 / Section 5
Most genes are shared across all animals. More than ninety-nine percent of human genes have a related copy in the mouse. As one examines animals that are more distantly related, the proportion of the genes they share decreases; however, despite about 500 million years of evolutionary separation, half the genes in the lowly sea squirt correspond to those found in humans. This remarkable conservation of gene structure is striking considering how much these animals differ in morphology, physiology, and behavior.
If they share so many of the same genes, why are different animals so different? Differences among species result largely from differences in the time and location of the genes’ expression. Let us consider our closest relative, the chimpanzee. Not only do chimpanzees and humans share nearly all of the same genes, but the DNA sequences of those genes also are very similar between the two species. Svante Pääbo sequenced three million bases of the chimp genome and found that chimps and humans differ overall by less than two percent at the sequence level. (See the Human Evolution unit.) Based on the low sequence divergence, Pããbo hypothesized that the difference between humans and chimpanzees was due mainly to how the genes were expressed in the different species.
To test this hypothesis, Pããbo compared the expression pattern of 20,000 human genes in humans and chimps. He found that while expression levels were similar in liver cells and blood, there were larger differences in brain cells. This suggests that the human brain has increased the use of certain genes compared to those same genes in a chimp. So, it not so much the sequence of the genes that is important, but how they are expressed to make the cell’s proteins that determines the unique characteristics of each organism.
6. Determining Gene Function from Sequence Information
Unit 1 / Section 6
Researchers have produced an enormous number of genome sequences from a variety of organisms. Publicly available databases, such as GenBank at the NCBI (National Center for Biotechnology Information), store many of these sequences. The databases have been a tremendous boon for comparative biology. The NCBI database stores not only the genome sequences, but also information about the function (if it is known) of the genes.
The NCBI can also identify unknown genes by comparing them with known genes in the database. One program commonly used for this purpose is BLAST (Basic Local Alignment Search Tool). Sequence similarity searching algorithms like BLAST are based on the premise that if two sequences are similar then they are likely to be homologous (that is, they share a common evolutionary ancestor). (See the Evolution and Phylogenetics unit.) Using this database, one can infer the function of an unknown gene by finding similar sequences of known genes and proteins. For example, suppose you were to use BLAST to search for sequences similar to a new gene. Upon viewing your results, you noticed that all the sequences with a high degree of similarity to the new gene belonged to a family of genes known to break down hydrogen peroxide. You could logically conclude, then, that this new gene encoded a protein with a similar function.
BLAST searches can be done at the nucleotide level; however, comparisons at the amino acid level provide much greater sensitivity. Therefore, unless one is particularly interested in the DNA sequence itself, it is better to search for genes using protein. If you have only raw nucleotide sequence data, computer programs can automatically translate the DNA into amino acids using all six reading frames (three frames from one strand and three frames from the complementary strand) before searching the protein database.
In addition to whole proteins, similarity searches can identify protein motifs. A motif is a distinctive pattern of amino acids, conserved across many proteins, which gives a particular function to the protein. For example, the presence of one particular motif in a protein indicates that this protein probably binds ATP and may therefore require ATP for its action.

Figure 4. BLAST search results.
The result of a database search is a list of matches, ranked from highest to lowest, based on the probability of a significant match (Fig. 4). The reported alignment scores are given “expectation values” (E), which represent the probability that a match with the reported score would be expected to occur by random chance. The smaller the E- value, the higher the assigned score and the less likely that the match was a coincidence. Some of the easiest results to interpret are very high scores (small E-values, low-probability), which usually result from two very similar proteins. Other easily identifiable results are very low scores, which indicate that the outcome is probably the result of chance similarity.
Search results also provide links (in blue) to a database page with information on each sequence similar to the query sequence. This page gives extensive information on the match sequence, including the organism it came from, the function of the gene product — if it is known — and references to journal articles concerning the sequence. BLAST results also provide the actual alignment results for nucleotides or amino acids between the query sequence and the match sequences.
7. The Virtues of Knockouts
Unit 1 / Section 7
Gene prediction programs have been valuable in the preliminary identification of genes; however, they have limitations. Unless the gene of interest is homologous to a gene of known function, the function is generally still not known. A biological approach to determining the function of a gene is to create a mutation and then observe the effect of the mutation on the organism. This is called a knockout study. While it is not ethical to create knockout mutants in humans, many such mutants are already known, especially those that cause disease. One advantage of having a genome sequence is that it greatly facilitates the identification of genes in which mutations lead to a particular disease.
The mouse, where one can make and characterize knockout mutants, is an excellent model system for studying genetic diseases of humans; its genome is remarkably similar to a human’s. Nearly all human genes have homologs in mice, and large regions of the chromosomes are very well conserved between the two species. In fact, human chromosomes can be (figuratively) cut into about 150 pieces, mixed and matched, and then reassembled into the 21 chromosomes of a mouse. Thus, it is possible to create mutants in mice to determine the probable function of the same genes in humans. Genetic stocks of mutant mice have been developed and maintained since the 1940s.
One goal of the mouse genome project is to make and characterize mutations in order to determine the function of every mouse gene. After a particular gene mutation has been linked to a particular disorder, the normal function of the gene may be determined. An example of this approach is the mutated gene that resulted in cleft palates in mice. The researchers found that the gene’s normal function is to close the embryo’s palate. An understanding of the genetics behind cleft palate in mice may one day be used to help prevent this common birth defect in humans.
8. Genetic Variation Within Species and SNPs
Unit 1 / Section 8
A polymorphism, the existence of two or more forms of sequence between different individuals of the same species, can arise from a change in a single nucleotide. These single nucleotide polymorphisms(SNPs) account for 90 percent of all polymorphisms in humans. The number of SNPs between two genomes provides a measure of sequence variation; however, the variation is not uniform over the genome. About two-thirds of SNPs are in noncoding DNA and tend to be concentrated in certain locations in the chromosome. In addition, sex chromsomes have a lower concentration of SNPs than autosomes.
There are about three million SNPs in the human genome, or about 1 per 1000 nucleotides. SNPs are ideal genetic markers for many applications because they are stable, widespread, and can often be linked to particular characteristics (phenotypes) of interest. They are proving to be among the most useful human markers for studies of evolutionary genetics and medicine.
Not all SNPs, even when they are present in coding genes, lead to visible or phenotypic differences among individuals. Changes in the DNA sequence don’t always change the amino acid sequence of the protein. For example, a change from GGG to GGC results in no change in the protein because both codons result in a glycine in the protein. This is called a synonymous mutation (silent mutation) or silent mutation; non-synonymous substitutions do cause a change in the amino acid. About half of all SNPs in genes are non-synonymous and therefore can account for diversity between individuals or populations. Depending on the particular change in an amino acid caused by a nonsynonymous mutation, the resulting protein may be active, inactive, or partially active. It may also be active in a different way.
One well-characterized SNP exists in a gene in chromosome 6. Individuals with cysteine at amino acid position 282 are healthy; however, about 1 in 200-400 Caucasians of Northern European descent possess two copies of that gene where the amino acid is tyrosine instead of cysteine. Due to this one change, these individuals have a disease called hereditary hemochromatosis. People afflicted with this disease accumulate high levels of iron, which causes permanent damage to the organs, especially the liver. About ten percent of these individuals carry only one copy of this mutation; they are heterozygous and are carriers of the disease. A genetic test for hereditary hemochromatosis is available, which can detect the SNP. If the disease is found, medical professionals can then determine whether the person is homozygous or heterozygous for this allele. Another example of a single SNP that has a dramatic effect is the one that leads to sickle cell anemia. (See the Human Evolution unit.)
9. Identifying and Using SNPs
Unit 1 / Section 9
In order to identify SNPs, nucleotide sequences of two or more genomic regions must be aligned so that the polymorphisms are apparent. Sequence alignments are easy when the sequences are similar, but can be very difficult when there are many polymorphisms. The alignment of two sequences is determined by a program that compares the two sequences, nucleotide by nucleotide. For multiple sequences, the program continues the same type of pairwise alignment for all possible pairs. The result is a pairwise distance matrix based on all possible alignments of any two sequences. This matrix is then used to construct a phylogenetic tree that predicts how closely related two sequences are, based on their similarity. The program then uses this information to align the sequences, again in order of their relatedness. This is the method used in a program called CLUSTAL. A typical output from CLUSTAL is shown in Figure 5.

Figure 5. CLUSTAL data
Most SNPs have no effect on an individual, so what use are having maps of them? SNPs appear to cluster in blocks called haplotypes. Grouping individuals that share a particular haplotype is called haplotyping. Because these particular sequences of SNPs on a chromosome are inherited together as blocks, they can be used to distinguish individuals and populations. What good is haplotyping? One can determine what specific diseases or other traits are associated with different haplotypes. In most cases, there are much fewer haplotypes than SNPs. Although it is the SNPs that actually cause disease, looking for changes in one SNP out of millions in the genome is not practical; looking for a particular haplotype is much easier.
An example of the value of haplotype comes from research on Crohn’s disease. Crohn’s disease is a chronic inflammatory disease of the digestive tract that tends to cluster in families. Researchers identified a haplotype on chromosome 5 that correlates with the disease. This region of the chromosome contains genes involved in immunity; these genes then may be important in other inflammatory diseases, such as lupus or asthma.
10. Practical Applications of Genomics
Unit 1 / Section 10Genome sequence data now provide tools for the development of practical uses for genetic information. DNA is an invaluable tool in forensics because – aside from identical twins – every individual has a uniquely different DNA sequence. Repeated DNA sequences in the human genome are sufficiently variable among individuals that they can be used in human identity testing. The FBI uses a set of thirteen short tandem repeat (STR) DNA sequences for the Combined DNA Index System (CODIS) database, which contains the DNA fingerprint or profile of convicted criminals. Investigators of a crime scene can use this information in an attempt to match the DNA profile of an unknown sample to a convicted criminal. DNA fingerprinting can also identify victims of crime or catastrophes, as well as many family relationships, such as paternity. While we think of forensics in terms of identifying people, it can also be used to match donors and recipients for organ transplants, identify species, establish pedigree, or even detect organisms in water or food. (See the Evolution and Phylogenetics unit.)
An unusual application of DNA fingerprinting technology is a project of Mary-Claire King’s at the University of Washington. (See the Cell Biology and Cancer unit.) Although her research is primarily concerned with the identification of genetic markers for breast cancer, she also has a project to help the “Abuelas,” or grandmothers, in Argentina. In Buenos Aires in the 1970s and 1980s, children of activists “disappeared” during the military dictatorship. The children were placed in orphanages or illegally adopted when their parents were killed. Now King is using mitochondrial DNA, which is inherited only maternally, to reunite the children with their grandmothers.
The basis of many diseases is the alteration of one or more genes. Testing for such diseases requires the examination of DNA from an individual for some change that is known to be associated with the disease. Sometimes the change is easy to detect, such as a large addition or deletion of DNA, or even a whole chromosome. Many changes are very small, such as those caused by SNPs. Other changes can affect the regulation of a gene and result in too much or too little of the gene product. In most cases if a person inherits only one mutant copy of a gene from a parent, then the normal copy is dominant and the person does not have the disease; however, that person is a carrier and can pass the disease on to offspring. If two carriers produce a child and each passes the mutant allele to the child (a one-in-four probability), that individual will have the disease.
Several different mutations in a gene often lead to a particular disease. Many diseases result from complex interactions of multiple gene mutations, with the added effect of environmental factors. Heart disease, type-2 diabetes and asthma are examples of such diseases. (See the Human Evolution unit.) Many diseases do not show simple patterns of inheritance. For example, the BRCA1 mutation is a dominant mutant allele that leads to an increased risk for breast and ovarian cancer. (See the Cell Biology and Cancer unit.) Although not everyone with the mutation develops the disease, the risk is much higher than for individuals without the mutation.
Newborns commonly receive genetic testing. The tests detect genetic defects that can be treated to prevent death or disease in the future. Apparently normal adults may also be tested to determine whether they are carriers of alleles for cystic fibrosis, Tay-Sachs disease (a fatal disease resulting from the improper metabolism of fat), or sickle cell anemia. This can help them determine their risk of transmitting the disease to children. These tests as well as others (such as for Down’s syndrome) are also available for prenatal diagnosis of diseases. As new genes are discovered that are associated with disease, they can be used for the early detection or diagnosis of diseases such as familial adenomatous polyposis (associated with colon cancer) or p53 tumor-suppressor gene (associated with aggressive cancers). The ultimate value of gene testing will come with the ability to predict more diseases, especially if such knowledge can lead to the disease’s prevention.
Gene therapy is a more ambitious endeavor: its goal is to treat or cure a disease by providing a normal copy of the individual’s mutated gene. (See the Genetically Modified Organisms unit.) The first step in gene therapy is the introduction of the new gene into the cells of the individual. This must be done using a vector (a gene carrier molecule), which can be engineered in a test tube to contain the gene of interest. Viruses are the most common vectors because they are naturally able to invade the human host cells. These viral vectors are modified so that they can no longer cause a viral disease.
Gene therapy using viral vectors does have a few drawbacks. Patients often experience negative side effects and expression of the desired gene introduced by viral vectors is not always sufficiently effective. To counter these limitations, researchers are developing new methods for the introduction of genes. One novel idea is the development of a new artificial human chromosome that could carry large amounts of new genetic information. This artificial chromosome would eliminate the need for recombination of the introduced genes into an existing chromosome. Gene therapy is the long-term goal for the treatment of genetic diseases for which there is currently no treatment or cure.
11. Examining Gene Expression
Unit 1 / Section 11
Understanding the functions of genes depends on knowing when and in what cells they are each expressed. How can one measure the amount of mRNA transcribed from a gene in a particular cell type? The standard method uses a probe – a DNA sequence unique for that gene – which binds to the mRNA that has the complementary sequence. The more mRNA particular cell produces, the more mRNA that is bound to the probe, giving the probe an increased signal. Because cDNA is complementary in sequence to mRNA, it can also be used to measure the expression of a particular gene.
Organisms have so many genes in their genomes that studying the expression of all of these genes had been exceedingly difficult. Going from studying gene expression one gene at a time to examining expression patterns of a multitude of genes required new technology.

Figure 6. Microarray experiment
In the late 1990s the development of microarray chips allowed researchers to examine the expression of thousands of genes simultaneously. This allowed for a much broader perspective of gene expression than was possible when genes were analyzed singularly. Microarray chips are glass slides spotted with many rows containing tiny amounts of probe DNA, one for each of thousands of genes (Fig. 5). The target sample of interest, usually made from mRNA of a specific type of cell, is labeled with a fluorescent dye and added to the chip. If there is a match between the sample of interest and the DNA probe on the chip, the two molecules will bind to each other. Then, when exposed to a laser, the spot will produce a signal that will fluoresce. (Figure 5 describes this process in more detail.)
Scientists can use microarrays, a rapid and sensitive test, in a variety of experimental studies. Using microarrays, one can measure expression patterns of large numbers of genes in different cell types (such as cancer cells versus normal cells, or liver cells versus kidney cells). It can also be used to examine the changes in gene expression over time (for example, as an embryo develops), or changes in a given cell type under different environmental conditions (various temperatures, for instance).
12. Ethics
Unit 1 / Section 12
Possessing detailed knowledge about the genetic makeup of individuals raises several complex ethical quandaries. How confidential should genetic information be? How should privacy concerns be weighed against other interests? If genetic information related to disease genes should be as confidential as any other health-related information, should there be databases of detailed genomic information on individuals? Even without detailed genomic databases, thirteen genetic markers are sufficient for the FBI to identify every person except identical twins. Should this type of genetic information be stored on all convicted criminals; everyone arrested for a crime; or on every individual, regardless of his or her past? Who should have access to detailed genetic information if it becomes available? Should it be accessible to law enforcement officers, physicians, research scientists, employers and potential employers, or insurance providers?
Sir Alec Jeffreys, the scientist who first developed the technique of genetic fingerprinting in Great Britain, is a proponent of a DNA database that contains the genetic profile of every individual in that country. To provide anonymity, however, he suggests that the actual identity of each individual be kept in a separate database with high security. Only certain circumstances, such as a link to a crime, would justify the identification of the individual.
The NIH-DOE Working Group on the Ethical, Legal, and Social Implications (ELSI) has recommended that employers can request and use genetic information, but only to protect the health and safety of workers; such information must remain confidential. They also recommend that insurers cannot use genetic information to deny or limit health insurance coverage or to charge different fees based on this information. Overall, the focus of legislation should be to prevent discrimination of individuals based on genetic information.
In 1993, long before the human genome was completed, a committee of the Institute of Medicine of the National Academy of Sciences developed recommendations to prevent involuntary genetic testing and protect confidentiality. They concluded that the responsible use of genetic testing requires that individuals understand the tests, their significance, and their implications. Testing for diseases should be done only when individuals are capable of providing informed consent. This means not only that individuals must be informed, but that they also should understand the implications of that consent. Such informed consent requires an understanding of genetics by the public. Education in genetics must be increased to ensure that future generations have this knowledge.
Patenting of human genes is another ethical concern emerging from the human genome project. In order to be patentable under the U.S. Federal Patent Act, an invention must be “novel, nonobvious, and have utility.” In applying for a patent on a human gene, applicants generally claim that the patent’s holders will add to the utility of the natural gene by developing tests and therapies to fight diseases associated with that gene. Opponents of gene patenting think that patents will limit the ability of other scientists to do additional research on these genes.
Most patents are filed by private companies that plan to develop and market diagnostic tests and treatments that come from their research on a particular gene. These companies feel that, without a patent, they cannot afford to do the research that will lead to useful products. They argue they need the protection of a patent before they can invest millions of dollars in the development of new tests, drugs, and therapies. Some scientists counter that companies tend to patent genes even before they know what the gene does, so it is hard to understand how they can claim that they will increase the utility of such a gene. Making scientific data freely available, while still protecting the interests of private organizations that will provide the practical uses for the data, would be in the best interest of everyone.
Epilogue
The explosion of information coming from the sequencing of genomes has changed the landscape of biology. We now have tools to better understand the basis of disease and its prevention and control. These tools also allow us to design more effective drugs, and even understand the genetic relationships among all living things that make the universal tree of life. Acquiring the sequence was only the beginning.
End Notes
1. Venter, J. C. 1998. Testimony before the Subcommittee on Energy and Environment. U. S. House of Representatives Committee on Science. 17 June 1998.
2. Dulbecco, R. 1986. A turning point in cancer research: Sequencing the genome. Science 231: 1055-56.