





Rediscovering Biology: Molecular to Global Perspectives
Proteins and Proteomics Expert Interview Transcripts: Ned David, Ph.D.
 Founder, Syrrx, Inc.
Founder, Syrrx, Inc.
Founder of Syrrx, Inc., a proteomics company in San Diego that specializes in x-ray crystallography and determining protein structure for use in drug discovery.
Interview Transcript
Ned David, Ph.D., is the founder of Syrrx, Inc., a proteomics company in San Diego that specializes in X-ray crystallography and determining protein structure for use in drug discovery.
What is the difference between proteomics and genomics?
I would describe [the genome] as a more static entity. It’s not completely static but when you are fertilized and Mom and Dad each contribute their genetic material, you undergo relatively limited alterations in your genome as you mature. Your immune cells shift around and there’s some discussion that your neurons do a little bit of that, but for the most part, you’re kind of done at that point.
Proteomics, on the other hand, is an organic and ever-changing picture on a cell-by-cell and organ-by-organ basis. The proteins are really the workhorses of life-they are increased in concentration, decreased in concentration, modified, destroyed, all as a function of moment-to-moment need.
And that’s what life is about because we are not static entities. We are things that grow, get sick, reproduce, and we die. Understanding how these parts work together-when they’re created when they’re destroyed, and how that works on a time-by-time basis, as well as a cell-by-cell basis, is really the basis of understanding dynamic life. I think that studying genomics is studying more of the static blueprint and studying proteomics is studying the dynamic life as it’s operating. I think each has utility.
What is the “then and now” of your proteomics research?
Ten years ago it would have been impossible to be able to perform comprehensive analyses on collections of proteins from cells and compare them to another collection of proteins from. A series of technologies have made an appearance that have enabled us to actually be able to do this type of cataloging [including] improvements in mass spectrometry technology, improvements in the ability to manipulate very small volumes of material, and improvements in our ability to manage the information and to make it mineable so that you can actually query that information. It enabled us to dissect the dynamics of life as viewed through the lens of proteomics. Prior to this, the complexity of all the different proteins in a cell was something you couldn’t even really talk about. It would be like predicting the weather. But now with these tools, we’re actually able to extract order from the chaos and be able to make meaningful assertions.
How did researchers study proteins in the past?
In the past, the ability to study large collections of proteins was really limited. I remember the first time I saw anything remotely resembling that-it was in the early ’90s, at a protein society meeting. It was the first time I’d ever seen 2D gels coupled with mass spec and it was just fascinating that you could actually do this. Prior to that, it was outside the paradigm to study large collections and look for differences.
As a matter of fact, [research] on questions such as “can you identify a suspect in this disease state?” used very different methods. You’d try using molecular biology to look for phenotypes that would be presented in cells. You sometimes met with success, [but] more often than not met with failure. [This new proteomics] approach allows us to get a step closer to the actual “bad doers” in these disease states and thereby have greater likelihood of success.
How did you start the proteomics company, Syrrx?
I was a graduate student at Berkeley in 1998 and I had filed my thesis in April. I was working on membrane proteins-these are very biochemically, ill-behaved proteins in the sense that they have no interest in being well behaved biochemical reagents for crystallography. They do everything crystallographers hate. They are not soluble in water, you can’t get them in high concentration as a biochemical reagent. But I banged on it for about six years in our lab. Then, my boss, Ray Stevens, who’s one of the cofounders here, thought of [an innovative] idea. Because getting proteins from membranes is hard, [and] you can’t get very much of them, [he suggested to] use very small drops to do the crystallography in. He built this robot to go along with this project that I’d been working on for about six years.
At about the time I filed my thesis and wrote up the first paper on the subject, Ray walked into the lab one day and he threw down some pictures on my bench of these very small drops in which protein crystals had formed. I had been thinking of starting a company based on membrane proteins and he walked in and said, “You know we should start a company based on these small drops” and he turns around the walks out.
Even though I’d been thinking about starting a very different company, within about two or three minutes I’d changed my mind and decided we’re going to [create] a company that automates all the key steps in protein crystallography so that we can determine the shapes of proteins more quickly and with greater success.
In the old days, it took multiple decades to [figure out] the first protein crystal structure, myoglobin. Then, of course, new technologies appeared to the point where you could get a protein crystal structure in a year if you were pretty good or sometimes even shorter with a big group of people.
DNA sequencing used to be done with these horrible polychrome gels and radioactivity. I did that for years when I was an undergrad and a graduate student. But then ABI came along and built the 3700 [machine] that allowed us to sequence a human genome in one year. It was this new tool that allowed you to make DNA sequencing fast and cheap. We thought, why don’t we try to do the same thing for protein crystallography? The idea was to come up with a means to build all these new tools that didn’t exist anywhere else or before and use them to build a factory to enable this protein crystallography process to be automated.
The goal’s really not about solving lots of protein structures. What people really care about is solving the structures of the things that really matter-the high-value drug targets where no one’s been able to determine the three-dimensional shape of these [proteins]. Our automation allows us to make large numbers of little changes in the protein to find one version that [will] form a crystal.
What are some of the challenges of finding new drug targets?
The human body has lots of parts, biochemically, and these parts all interact. We only know a very small amount about what parts are interacting in the important places. To figure out the very right place to intervene-where if you just inhibit one thing that disease will go away-is very hard.
The whole effort behind genomics and proteomics is to try to identify these useful points of molecular interaction in disease states, and it turns out to be much tougher than anyone ever would have thought. Most of the targets that are getting nominated by genomics and proteomic efforts are [at a] very low validation state, meaning no one has any idea [whether it would work] to put a small molecule against any of these. The big challenge [that] we currently face as we try to pull ourselves out of medieval medicine into some sort of futuristic type of medicine where we really know where to go, is figuring out a way to pick [better drug targets] out of the morass of potential targets that we’re able to nominate.
This process, called “target validation,” is an art. It is not a science. It has scientific elements, but figuring out which ones are the high validation states takes a lot of work and ultimately can’t even be proven well in animals. We have a lot of examples of drug targets and drugs that work great in mice. We can cure cancer in mice wonderfully. But it turns out these animal models are not highly predictive necessarily, and as such, you have to ultimately get into humans to test some of this stuff.
What is the difference between working with a gene vs. a protein?
When you manipulate a gene, for example, if you’re making a construct that includes the nucleotide sequence of that gene, it’s something that’s very mechanical. It’s like carpentry. DNA is made out of four bases, so the overall charge-to-mass ratio is basically always the same. Mostly, they don’t ever surprise you when they move on gels.
Whereas proteins have 20 different amino acids and they fold up in these funny shapes — each one of them has different biophysics, so when you try to physically handle them as reagents they will do counterintuitive things on you. [Perhaps one] isn’t soluble anymore, or only soluble in certain pH’s.
Can you talk more about protein structure specifically?
If nature has a purpose, you can say that the purpose of using 20 different amino in a linear sequence is to allow nature to design complex three-dimensional shapes to be structural elements or to be catalysts for biochemical reactions. Nature really needs these scaffolds so it can program these three-dimensional shapes because it’s these shapes that confer function.
If you didn’t have these different elements to choose from, there’s no way you could program that type of three-dimensional complexity into a biological molecule. It’s the reason DNA couldn’t do this on its own. Whereas proteins with all their complexity — which of course makes them difficult to work with — are the ideal templates to [from which to] design complex life.
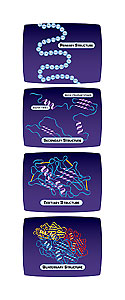
The primary structure of a protein simply [is] the linear amino acid sequence-you could write down on a piece of paper the primary structure of a protein. Of course, that is only the actual chemical information, and it has no structural three-dimensional information.
The secondary structure [of a protein] is comprised of signature folds that proteins typically take up: typically seen are things like alpha-helices, which look like corkscrews. Another secondary structural element is called a “beta-sheet” wherein the peptide backbone sort of traverses one direction turns around and comes back, hydrogen bonding across as it moves. And what you’ll find is that nature mixes and matches these secondary structural elements to generate overall static structures of the proteins-this overall structure is called the “tertiary structure.” [It’s this] part that becomes the real business end of what we work on-it’s the actual shape that confers the biochemical or structural function upon the protein.
There is a higher order of structure which is called “quaternary” wherein different subunits bind to and work in concert with other identical members or even a different protein. This quaternary structure describes the larger supermolecular assembly of the different protein subunits.
If you know the amino acid sequence of a protein, will you know the three-dimensional structure as well?
If you have a linear sequence, it will fold up into a 3D shape that is determined by physics. Therefore, it follows that if you have a computer you should be able to use math and physics to understand exactly what three-dimensional shape is going to be formed by a linear sequence-you should be able to design proteins and you should certainly be able to predict ones.
As it turns out, while that is theoretically true, [determining the] three-dimensional shape based on primary protein sequence is computationally very hard and it’s something that human beings are only now getting at. A lot of people get [the structure] wrong. In the future and perhaps in the next decade or two we’re going to be much, much better at this because it is determined by physics — there’s no mysticism here. An understanding of these rules will occur and getting the computational power to actually apply these rules to the system will become more broadly available.
What is the importance of a protein’s three-dimensional structure?
Why did nature evolve proteins to do this? Why didn’t it choose to use DNA or RNA as a means to be structural material or catalytic material? It really has to do with the diversity of chemical structures that you can call upon.
It allows us to have a metabolism which is a really underrated thing — it’s really [a great innovation] that we’re able to actually eat things around us which are really heterogeneous stuff and we are able to convert it over into things we can use. It’s astonishing. This is all enabled by the fact that these proteins have great substrates for evolution-essentially proteins evolved for evolvability. Very subtle changes that you can make in a protein in one location can very subtlety tweak a shape in an active site so as to allow you to gain a type of specificity you need to make a biochemical pathway work.
[Here are] some specific examples of a 3D structure doing things biochemically: We’ve known that enzymes, which are protein catalysts, are able to accelerate the rate of a chemical reaction, by stabilizing a transition state that appears only for a short moment in solution. It [is able to] lower the barrier energetically to perform the reaction. It might lower it a few orders of magnitude, so instead of it taking a thousand years to perform a reaction it might get it down to five minutes, because it lowered the energy of this transition state by basically grabbing it from solution using it’s 3D shape, stabilizing it energetically and thereby letting the reaction take place.
Why is it so difficult to resolve a protein’s structure?
What makes our job difficult in terms of working with proteins is because nature did not evolve them to form crystals. And we need to get them to form crystals for us to do rational drug discovery using their shape. So what [do we] do? The answer is automated processing. We [thought that if we could] make hundreds or even thousands of genetic variants of a given protein [we might find a form that was] easier to work with [because it would] form a crystal.
This approach was very unorthodox because [we] were modifying nature. [We were using] essentially an evolutionary approach to introduce changes into the protein to make it “well behaved” and we call this approach “combinatorial protein expression.” This approach has allowed us to get versions of proteins that form crystals where no one else has succeeded.
Syrrx is working on the DPP4 protein: can you talk about that?
DPP4 is a very odd enzyme. It pokes out of your cells, so it’s actually not inside the cell. Its purpose is to degrade the circulating hormones in your blood. One of these hormones is responsible for elevating insulin production, and so we realized that if we could inhibit the degradation of this thing it would be the equivalent of taking oral insulin stimulator.
We [knew we needed to] get the structure of [DPP4]. [Then we had to find] the structure of a variety of other compounds that people have created [that] bound to this structure. [Our goal was to] understand the best way to design a drug based on everything else people have done in the industry-essentially stand on the shoulders of giants to make the drug.
We had a tall order, because no one had ever solved [this] structure before. [We eventually] got a structure [at] very high resolution so we can see many of the atoms. We’re now generating on a weekly basis new structures of the protein DPP4, bound with small molecules that we have designed that can become starting points for drug discovery.
What is the step-by-step process of finding a drug target?
We begin with DPP4. We take the gene and we design constructs where we include different amounts of the “business end” of this gene product, this being the part that is responsible for performing the catalytic function of degrading these circulating hormones. We make a variety of these different constructs that include the core bit of the gene plus what we call “spinach” attached to either side of it that help purification or crystallization. The constructs are circular pieces of DNA that we’re able to move into cells to instruct those cells to produce the DPP4 protein.
These DNA constructs are moved using viruses into insect cells and these insect cells are then grown up to produce biomass. We then take these infected insect cells and crack them open [to] extract the protein. We then purify the protein using a series of robots, as well as humans.
We then take the protein that we’ve purified and concentrate it so we get a lot of protein in a relatively small volume. This protein is then handed to our crystallization robot [which] then proceeds to set up a very large number of very small crystallization drops. We do this in a drop volume that is anywhere from 50 to a hundred times smaller than conventional crystallization drops. We call [these] “nano drops.” The crystals then grow in these nano drops. A series of robots then looks for the protein growing and tries to find which drops have the crystals. A computer then finds the crystal and flags a [person]. A [person] comes in, harvests that crystal, freezes it in liquid nitrogen, and we drive it up to Berkeley where we put it in an x-ray beam that is generated as a byproduct of a synchrotron. We do this at the Advanced Light Source in Berkeley.
The x-rays generated by this synchrotron are shot down a very long tube of optics, ultimately to be blasted on top of the crystal. The crystal diffracts the x-rays to form a pattern. This pattern is a series of spots. We then turn the crystal with respect to the beam to collect other images and we’re able then to take these images to determine the three-dimensional shape of DPP4.
What do you do after this x-ray crystallization process?
There’s a lot of data that we generate from this. We have the computationally intensive task of using this collection of spots to get the three-dimensional picture of the protein. We use a variety of computer programs to convert the spots into a three-dimensional picture of a protein.
So after you know the 3D structure of a protein, how do you design a drug?
At this point we really get down to the business of making drugs. And the goal when you’re doing rational drug design is to use the shape of this protein combined with information about how a small molecule binds to it to design a drug to fit snugly much like a key into a lock. We’ll look at this protein structure, and we will then visualize the drugs bound by actually co-crystallizing the drug with the protein and then visualizing it.
[A group of people] will look at these and they will suddenly start just pouring out ideas of design. As a matter of fact, most of the key ideas that become the basis for the drug that ultimately goes into the clinic and into humans oftentimes come out in the first few hours after looking at the protein structure. It’s really the roadmap toward the design of the ultimate drug.
During these design sessions, the people will put on 3D glasses, sit around a computer, and physically stare at the active site, seeing the drug bound to the target and say, “You know what we ought to do? We should take this piece from here and take this piece from this other complex and fuse them together and try that and see what happens.”
And then the chemists are told to go make that. A compound takes a little while to make and then that’s part of the next iteration for company-crystallization bound to the target.
What happens when you find good leads for potential targets?
The chemists then prioritize the compounds based on how difficult or hard they are to make. It is also based on the likelihood of success; that is, based on goodness of perceived fit. Then the last parameter is if we are going to be free to operate. Does anyone have a patent on that?
Now we have to go synthesize them. So the chemists come up with synthetic means to make all of these chemical pipedreams that we had based on staring at the structure. Some of these things are easy for them to make and other ones are complete bears. Oftentimes even though there’s a good idea, we can’t make it because it would take six months and if something instead takes six days and we can get the next step in the iteration process we’ll just go that route.
[After the compounds are made]-often a dozen or so at a time-they then get sent to screening and they will be characterized for how tightly they bind. Oftentimes we’re right that this one’s the best one; this one’s the worst one. Sometimes we’re wrong. But, you know, with the structure guiding you, you’re right quite often.
How is healthcare changing because of proteomics?
We are at the edge of massive change in how we think about disease and certain aspects of what I called “managed mortality.” This is not well appreciated. The changes are sufficiently gradual that people are not tied into what’s happening.
[For instance,] the drug Gleevec that got approved last year was one of the first [drugs] that is a selective, rationally designed inhibitor of a signaling protein called the “kinase” and it was a specific target that was picked because it was modified in a particular cancer state, so it was a pretty high validation because it only appeared in this very rare cancer called CML.
If you got CML, it was a death sentence and [the] scientists at Novartis said, “Let’s make an inhibitor of this.” They designed it, they put it through clinical trials, and if you take this drug your cancer goes away. And it’s relatively nontoxic. I mean this is a remarkable achievement. Now you get Gleevec resistant mutants and people are now dealing with that problem, but the fact is that this worked.
Imagine ten years from now where we have a dozen examples of that, not one. How are we going to think differently about the options we have to manage our mortality? I think that these are the types of things we’re going to see a lot of in ten years and even more in 20.
What other changes do you see in the future for this field?
We’re now in the process of watching drugs move through clinical trials that will fundamentally alter how humans think about their mortality. There are going to be more choices that never existed before for how one interacts with diseases, and we are going to be able to make choices about how we manage our dignity, and it’s going to be interesting from a policy perspective. Who’s going to pay for that? It’s going to be interesting to observe people that choose to participate or not participate in those options and it’s also going to be interesting to see how far we can push it to actually make these options available. What things will we not be able to alter about ourselves and what things can we alter?
Are there ethical issues in proteomics?
A lot of the diseases we work on are clearly diseases of privileged countries and we do that out of necessity. We couldn’t be in business, we couldn’t be doing research if we were working on tropical diseases, for example.
For example, malaria kills more people than I think any other disease, but you’d be hard-pressed to identify a pharmaceutical company with a large malaria program because the people that have malaria have dirt floors and they don’t have debit cards and BMWs and as such, there’s a definite bias toward working on diseases that affect the world’s privileged.
How do I feel about the ethics of that as a researcher? I think that it becomes an issue of the minimization of evils that you’re engaging. For me, inaction, decision not to participate in research to increase human dignity would be unethical and the options then left to you are do you do it and operate within the economic rules imposed on you, or do you choose not to do it. The only ethical decision is to participate and I think that anyone who would choose not to participate by virtue of the inequity should examine the ethics of their decision.
Where do you hope to see the field of proteomics going in the future?
My hope is that we are able to use these new technologies to essentially increase the dignity with which humans can live. There are so many diseases and problems that we suffer from on a day-to-day basis. Biology is essentially a series of mechanical interactions with proteins and nucleic acids, and it’s a medium that we can manipulate if we understand it. [It should be] done ethically and with an eye towards increasing human dignity rather than taking it away. I think that’s where I’d like to see the ultimate fruits of what we’re doing take us.