


Rediscovering Biology: Molecular to Global Perspectives
Cell Biology and Cancer Expert Interview Transcript: Robert Weinberg, Ph.D.
 Member, Whitehead Institute
Member, Whitehead Institute
Weinberg is a founding member of the Whitehead Institute for Biomedical Research. Weinberg and his colleagues discovered the first human cancer-causing gene, Ras . Weinberg has written and edited five books and more than 290 articles.
Interview Transcript
Interview with Robert Weinberg, Ph.D. Weinberg is a founding member of the Whitehead Institute for Biomedical Research. Weinberg and his colleagues discovered the first human cancer-causing gene, Ras. Weinberg has written and edited five books and more than 290 articles.
What have we learned about cancer since Nixon’s war on cancer?
When Richard Nixon launched his war on cancer in 1970, there was a widespread belief that viruses were the cause of most kinds of human cancers and in fact, there was an enormous research program that was launched in the years that followed trying to track down these human cancer viruses.
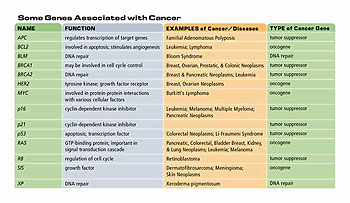
In truth, that program largely failed because many kinds of human cancers have no viral associations at all. However, there was an enormous wealth of information that came from that almost in an unintended way, which revealed that there was another class of agents-cellular genes-that are indeed very important for creating the genesis of human cancers. These cellular genes came to be known as “proto-oncogenes,” and are involved in regulating positively normal cell proliferation. They stimulate normal cells to grow when the tissue in the body needs them to grow.
In [1979], Harold Varmis and Mike Bishop first discovered this class of genes. These proto-oncogenes can become mutated when they reside in cells, and once they become mutated they can now be runaway growth-promoting genes. So they go from promoting a low level, slow amount of cell proliferation, to a very high level-they drive the cell to proliferate inexorably. And this unrelenting proliferation is one of the hallmarks of cancer cells.
[So] we have learned that cancer is ultimately a disease of genes, that there are damaged genes that reside in the genomes of cancer cells that push these cells to grow. There are [also] damaged genes in the genomes of cancer cells that are normally responsible for holding down cell proliferation and these genes are lost. There are [also] a variety of other genes that conspire with these growth-controlling genes to create the unregulated runaway proliferation of cancer cells.
No longer do we need to talk about cancer as being a mysterious entity. We now understand with great precision in molecular terms the agents that are responsible for inducing normal human cells to grow like malignant cancer cells.
How did our understanding of cancer change so that we were able to come up with a drug like Gleevec?
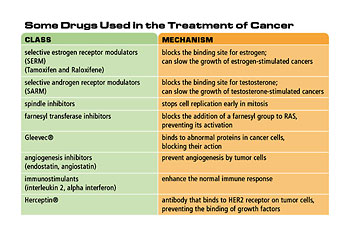
The fact that we could associate runaway cancer growth with certain distinct genes and therefore proteins meant that one suddenly had targets within cancer cells that could be attacked in principal by drugs.
The drug recently developed by Novartis called Gleevec is one example of that. That drug attacks an enzyme known as tyrosine kinase that is deregulated in chronic myeloid leukemia. The drug prevents that enzyme from firing properly and, as a consequence, cancer cells are no longer given the growth impetus that they would normally enjoy when this tyrosine kinase fires uncontrollably.
Hence, one now enters the era of rational drug design, where one can design and specifically target drugs to specific molecular entities inside cancer cells such as this tyrosine kinase. It’s no longer a “hit and miss” undertaking.
Was there one big question everyone was trying to answer?
The speculation in the beginning of the 1970s was that somehow there were damaged genes inside cancer cells, and those damaged genes were in some way responsible for malignant cell proliferation-but that was only a speculation. During that decade, one nailed irrevocably and irrefutably the fact that there are some damaged genes inside the genomes of cancer cells and that those damaged mutant genes are indeed directly responsible for causing the runaway proliferation of cancer cells. That was the major victory, the major glory, of that decade of cancer research.
Why would you say cancer has been and continues to be such a challenging disease to figure out?
The challenge in trying to understand cancer cells is that essentially they’re not that much different from normal cells. We can imagine that a normal human cell has between 28 and 30,000 genes and a cancer cell has the same set of genes except maybe five, six, seven, or eight of them are mutated, and therefore we’re looking from very minor subtle genetic differences in a very large genetic haystack.
What’s your big dream in this area?
For someone like myself who’s interested in understanding the causes, the origins of cancer, the ultimate goal is to be able one day to draw a circuit diagram of the cell to be able to visualize how all the molecules inside a cell talk to one another and collaborate and synergize to create the complex programs of cell proliferation that operate both in normal cells and in cancer cells.
We’re still a decade or two away from being able to do that, but once we do that we will, for the first time, really understand how and why genes and proteins cause cells to behave normally or abnormally.
How did molecular biology make a big impact in this field?
The fact is that the origins of cancer lie in genes. Another fact is that before about 1977-78 we couldn’t really work with those genes because we didn’t have the tools for isolating specific human genes-plucking them out of complex cellular genomes.
The advent of molecular gene cloning came just at the right time. It came just when we needed to begin to pull these genes out of cancer cell genomes and study them, isolate them, and be able to understand their sequences and how they operated. Without the revolution in gene cloning, we wouldn’t be anywhere near where we are today. We would still be stuck in the quagmire that surrounded us in the mid-1970s.
Could you tell me the story about the proto-oncogenes?
In 1975-76, Harold Varmis and Mike Bishop in their laboratory isolated a very interesting cancer-causing gene-an oncogene from a chicken virus-that caused cancer in chickens. They thought that this gene was specific for the cancer-causing virus, but then they looked further and they discovered that a very similar copy or version of that gene was present in normal cellular DNA and from that they deduced for the first time that within normal cellular DNA there must lie certain kinds of genes that when they become corrupted or mutated, they take on/acquire the power of causing cancer.
They called the normal genes “proto-oncogenes” and the cancer-causing “oncogenes.” The term “proto-oncogenes gene” signifies the potentiality within a gene of eventually becoming an oncogene, of becoming converted from a normal benign growth-promoting gene into an agent of malignant change.
The proto-oncogenes are present in our genomes and in the genomes of all of our ancestors for the last 500 million years. They are there because they play essential roles in controlling normal cell proliferation. Without the proto-oncogenes, embryos wouldn’t be able to develop and adult tissues would not be able to be maintained. However, the price of carrying these proto-oncogenes with us in our genomes is that occasionally they become damaged and mutated and convert into oncogenes and thus become converted into agents for causing cancer.
Can you discuss your work with Ras and how that discovery came about?
In the late 1970s, we began to look at cancer genes we imagined resided in human tumor cells. The human tumor cells had no viral associations. There were no viruses that came in and infected them and converted them into cancer cells. They had some kind of other origin.
We started looking at cancer cells that had been converted into cancer cells through exposure to chemical carcinogens; first mouse cells and then human cells. The human cells came from a human bladder carcinoma from cigarette smokers whose urinary bladder is exposed to high concentrations of mutagenic chemicals through the years that they smoked.
When we took DNA from the genome of a human bladder carcinoma cell and transferred it into a normal cell, the normal cell began to grow in a malignant or transformed fashion. And thus that indicated that the information for being cancerous resided in the DNA molecules that we had transferred from the human cancer cell into the mouse cell. When we isolated the human cancer-causing gene, the human bladder carcinoma oncogene, we found that it was a relatively small gene and lo and behold a normal version of that gene resides in all normal human DNA. The difference between the normal human gene and the cancer-causing bladder carcinoma gene was very subtle. Out of 6,000 bases of DNA sequence, there was only a single base, a single nucleotide, that was altered and that single nucleotide alteration, so-called “point mutation,” was sufficient to convert a normal cellular proto-oncogenes gene into a virulent oncogene.
Besides point mutations, what other ways can a gene can transform into an oncogene?
There are many ways by which a normal proto-oncogene can be converted into an oncogene. Point mutations are the way by which, for example, Ras genes become converted from proto-oncogenes to oncogenes. Other genes undergo a process called gene amplification where there may be two copies of the gene present in normal cells and 20 or 50 or a hundred copies of the gene, often a growth-promoting gene, present in cancer cells, so gene amplification is very important.
Also, chromosomal translocation may become important. In this mechanism, a region from one chromosome is inadvertently stuck together with a region from another chromosome and the resulting fused gene now begins to fire uncontrollably simply because the two genes have now been juxtaposed in a fashion that is most unnatural-this prevents the gene from firing normally. There yet are other mechanisms that are involved but these three are really the most important.
What was the significance of the discovery of Ras in cancer research?
The discovery of the point-mutated Ras oncogene provided the first specific proof that there were in fact mutated genes in human cancer cells. Until that point in time, it was only speculation that gene mutation was causative for human cancer. The discovery of a gene which was not only mutated, but carried the power of converting a normal cell into a cancer cell, provided the conclusive proof that the origins of cancer could be traced directly to mutated cellular genes.
How and why are genes mutated on the external level?
In the case of the bladder carcinoma oncogene-the point mutator oncogene that we isolated in 1981-82-the history of its origins was pretty clear. The bladder carcinoma from which we cloned this gene was isolated from a 55-year-old man who’s smoked since he was about 15 years old. During those 40 years, the carcinogenetic chemicals, the mutagens from the cigarette smoke, had been passed from his lungs into his urinary bladder on route to being excreted.
While these mutagenic chemicals resided in his bladder, they attacked genes of cells lining the walls of the bladder. They attacked those genes randomly and willy-nilly creating mutations here, there and everywhere. In the great majority of cases, those resulting mutant genes had no effect whatsoever on the process of cancer formation, but on very rare occasions, maybe one out of ten or a hundred million point mutations hit a jackpot. It hit right into the Achilles heel of a Ras proto-oncogene, thereby converting it into an oncogene. Now that cell with a mutant oncogene and all of its decedents had a great proliferative advantage over all the other cells in the bladder. That cell could grow much more rapidly than all other cells lining the bladder, and that cell became the progenitor of the large number of cells that were ultimately discovered to form the bladder carcinoma in that 55-year-old man.
What else besides chemical exposure can cause gene mutations?
We understand some of the sources by which genes become mutated, but hardly all of them. We know clearly that cigarette smoke is responsible for maybe a third of all human cancers-there are large numbers of mutagenic highly potent chemicals present in the combustion products of tobacco that are responsible for mutating DNA.
We know that ultraviolet radiation is highly mutagenic and is responsible for basal cell carcinomas and melanomas. We imagine that cooking meats at high temperatures generates all kinds of mutagenic compounds that are responsible for contributing to colon cancer. But for many kinds of tumors in the human body, it’s not exactly clear what the mutagens are that are responsible for creating mutant genes and ultimately generating cancers.
Can you talk about Ras as a G protein and the signal transduction pathway?
We know that the way by which a normal cell receives signals for proliferation is that on the surface of the cell there’s a growth factor receptor, which pokes out of the cell like an antenna that receives the signal that it’s bound EGF, epidermal growth factor, which floats through the medium. The receptor receives that signal and then sends the signal into the cell that informs the cell of this encounter and the cell ultimately responds by beginning to proliferate.
There’s a whole chain of command. It’s often called a “signal transduction cascade” or a “pathway” that flows below the epidermal growth factor receptor or other growth factor receptors that ultimately reach into the heart of the cell, the nucleus. It’s a long complex bucket brigade and one of the partners in that bucket brigade is the protein called Ras. Ras receives signals from upstream the bucket brigade and in response to those signals, it passes them on further down. This receipt and further transmission is sometimes called the process of signal transduction.
The Ras protein exists in a very interesting way. It acts as a binary switch, either it’s off or it’s on. Normally it’s off and it’s not emitting any signals. It’s just sitting there like a bump on a log doing nothing. But if it receives an upstream signal from some kind of growth factor receptor that informs the cell interior that the cell has encountered a growth factor, then the Ras protein is kicked into the On position and it stays in this On position for several seconds. While it’s on the On position it will send out signals to the interior of the cell pushing the cell to proliferate. And then after a couple of seconds the Ras protein will shut itself off. It’s like a light switch that is turned on but automatically shuts itself off after several seconds ensuring that there’s only a brief burst of signaling or light coming out the downstream end of the protein.
What we found in the early 1980s, was that the oncogene protein, made by the Ras oncogene, is kicked on into the On position like its normal counterpart and sends out all of these growth signals. However, the mechanism by which the Ras protein shuts itself off and puts itself in the down position is defective. And what happens, therefore, is that the Ras oncogene protein is kicked into the On position, sends out signals, and stays in this On position not for seconds but for minutes, hours, maybe even days, flooding the cell with growth stimulatory signals which are obviously quite inappropriate, at least in normal cell physiology. Therefore a cell carrying a Ras oncogene and making a Ras onco-protein is pushed to grow inexorably instead of being exposed to brief bursts of growth stimulatory signals. That is one of the prime attributes of cancer cells-this unrelenting inexorable growth.
Can you talk about the multi-hit nature of cancer?
At one point, we thought that if a cell acquired a single point mutation and through that acquired a Ras oncogene it would start growing as a cancer cell but that view was naive. We should have known better. The fact is that it takes a large number of alterations in a normal cell before it will truly grow as a cancer cell.
In the case of the bladder carcinoma, the Ras oncogene and its creation represented only the first step of probably four, five, six subsequent steps that occurred during the long multi-step evolution that intervenes between the inception of the formation of a cancer and the ultimate appearance of a clinically detectable tumor. In most kinds of human cancers, we imagine 40, 50, 60 years intervene between the inception and the ultimate appearance of the tumor.
Obviously, certain lifestyles can compress that timescale. For example, if one smokes cigarettes and floods one’s body with high numbers of carcinogens all day then the rate with which new genes are mutated is enormously increased and the time required for a cancer cell to acquire all of these different mutant alleles is compressed from 40, 50 or 60 years maybe down to 20 or 30.
So does that explain why it tends to be a disease of older people?
In effect, cancer is largely, not exclusively, a disease of older people simply because most kinds of cancer require so many years to occur. The formation of a single kind of human cancer represents the convergence of a series of unlikely events, each of which might only happen once in a decade in a given tissue. Therefore cancer is largely a disease of people who’ve lived long enough for all of these hits, all of these alterations to take place.
What are some of the body’s defenses against these mutations?
The adult human body has between three and five times, 10 to the 13th cells in it. That means between 30 and 50,000 billion cells in a single body. If you ask how many different cell divisions occur during an average human lifespan it’s on the order of 10 to the 16th, a staggeringly large number, and each one of those cell divisions represents an opportunity or a danger for something to go awry and as a consequent for mutant genes to accumulate.
As such, during our evolution, we’ve had to acquire a large number of potent anticancer defense mechanisms that ensure that in spite of this enormous risk of cancer tumors actually don’t appear that often. If one leads a virtuous lifestyle and doesn’t smoke and eats a reasonable diet the chance of one dying of cancer is probably about 10%, 1 in 10.
Imagine, then, in each of us goes through 10 to the 16th cell divisions in a lifetime and only 1 in 10 of us actually succumbs to cancer. That means that these anticancer defense mechanisms are so effective that they reduce the risk of cancer to 1 in 10 to the 17th cell divisions. I think that’s stunningly successful.
How did research on the cell cycle help to advance the understanding of cancer?
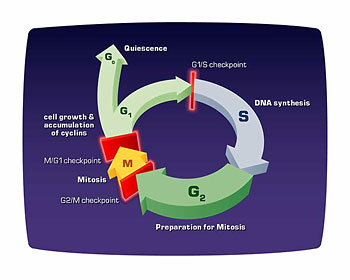
We always knew that cells could grow and divide, at least we’ve known that for the last 150 years, but what we really didn’t understand is the complex program that makes all that possible. The fact is, after one cell divides into two daughter cells, now each of those two daughter cells begins to grow, and ultimately the contents in each of these two daughter cells doubles so that each of these two daughter cells can, in turn, become the mother of two more cells, two grandchildren if you will.
And this complex program of accumulating double the amount of constituents inside a cell is what’s known as the “cell cycle.” In fact, it’s been much easier to study the cell cycle in primitive nucleated cells like yeast cells than it has in mammalian cells, so much of the pioneering work came from studying the way baker’s yeast grow and divide.
From these studies of the genetics of baker’s yeast, we began to understand the complex molecular machinery that choreographs this complex program whereby a cell goes through this cycle of growth and division. Included among the components of the cell cycle is a phase where the cell actually copies and replicates its DNA, thereby doubling it, and another phase where the cell divides in what is known as mitosis. And each of these phases is done with great precision so that if anything should go wrong the cell stops and repairs or tarries until all the defects are cured before it proceeds to the next step in the cell cycle.
If that weren’t there then cells would rapidly destroy themselves because of their ineffective way of replicating themselves exactly in a fashion that is required so that daughter cells are true replicas of the mother cell from which they derived.
The direct relevance of the cell cycle to understanding cancer only became clear when a number of groups began to study another group of genes that came to be called “tumor suppressor genes.” The proto-oncogenes and oncogenes are involved in pushing up cell proliferation. These tumor suppressor genes in the life of the normal cell are interested in damping down cell proliferation.
Often we find in the genomes of cancer cells that these tumor suppressor genes had been deleted or in some other way inactivated. So one makes the frequent metaphor that a cancer cell resembles a runaway car that has a stuck accelerator which forces forward uncontrolled growth-an activated oncogene, a growth-promoting gene. At the same time, cancer cells lack brake linings, the inactivated tumor suppressor genes with which they might slow themselves down.
Tumor suppressor genes are normally responsible for shutting down cell proliferation and in most kinds of cancer cells, these two kinds of alterations conspire with one another to give runaway growth-activation of an oncogene or inactivation of a tumor suppressor gene is not enough.
What is the role of tumor suppressor genes in the cell cycle?
With the discovery of tumor suppressor genes, we came to realize how important the cell cycle is. One of the most intensively studied of these tumor suppressor genes is the retinal blastoma (RB gene). If one inherits a defective version of the RB gene from one’s parent, then one has a very high risk of getting the rare eye tumor of retinal blastoma.
In fact, the protein made by the RB gene is a controller of the advance of the cell through a specific phase of its cell cycle. It’s a breaking mechanism so that if a cell advances through the G1 phase of the cell cycle, the time between mitosis and DNA replication (S phase), the RB protein will stop proliferation if conditions are inappropriate. In the absence of the RB protein, cells proliferate uncontrollably because this braking or stoppage mechanism is no longer in place to prevent or preclude inappropriate advance through the cell cycle.
Can you talk about Henry Harris’ work to fuse two cells together expecting cancer to take over?
The notion at least from work on tumor viruses was that cancer is a dominant phenotype and normal growth is a recessive phenotype. In fact, when Henry Harris, a biologist at Oxford University, tested this theory by fusing cancer cells with normal cells, the resulting hybrid cells actually grew like normal cells. This turned the whole field of cancer genetics on its head because it suggested that cancerous growth is in fact recessive and normal growth is in fact dominant.
As we learned later, the reason for these unexpected behaviors of these hybrid cells [is that] the tumor suppressor genes play a very important role in enabling cancer cells to grow uncontrollably. Therefore, cancer cells lack alleles of tumor suppressor genes, so those are obviously recessive alleles. Normal cells contain copies of well-functioning tumor suppressor genes which slow down their normal proliferation and when the two cell types are fused with one another they create a hybrid genome. The intact, normally functioning tumor suppressor genes donated by the normal cells are able to suppress the proliferation of the resulting hybrids explaining Henry Harris’ result coming from the hybridization of cancer cells and normal cells.
Was his experiment one of the things that led us to find the tumor suppressor gene?
The discovery of tumor suppressor genes was initially provoked by studying this rare eye tumor, retinal blastoma, and the genetics with which it was inherited. Alfred Knudsen, a geneticist in Texas and in Philadelphia, was able to measure the rate with which this rare eye tumor appeared in children from families where retinal blastoma was prevalent. At the same time, he studied retinal blastoma in families where there had been a history of retinal blastoma. And what he postulated on the basis of these studies was that there are actually two genes involved with retinal blastoma. If one inherits one defective copy of the RB gene, the retinal blastoma gene, then one has a predisposition to losing the other copy, he suggested, and when both copies are lost all hell breaks loose and a cell begins to grow uncontrollably.
In children who inherit two good copies of the RB gene, one copy is first lost as a somatic mutation and then the other one-now the child has a cell that lacks both copies of the gene and that cell grows uncontrollably. This suggested that the creation of a cancer cell depended on the successive inactivation of two copies of a growth suppressing gene which we came to call a tumor suppressor gene.
Can you talk about p53 and why it is studied so much?
The p53 gene is mutated in as much as 50% of all human cancers and it’s probably the case in the remaining 50% that the p53 protein is also compromised in its functioning. Why is it so important? Why is it so frequently altered?
Normal human cells have an alarm mechanism that triggers them to stop growing or even to die if things are amiss. If they experience too little oxygen, if their metabolism gets messed up and unbalanced, if they sustain massive damage to their genomes, if they receive too many growth-promoting signals, any one of those can suffice to trigger an alarm signal. This alarm signal, in turn, can cause the cell to stop growing, often irreversibly, or cause the cell to die, the process of program cell death that we often call “apoptosis.”
At the middle of this alarm system circuitry lies the p53 protein. The p53 protein is essential and a central clearinghouse that receives all kinds of signals from monitoring systems scattered through the cell. [These signals] tell p53 that things are normal or abnormal, and in response to certain kinds of signals of abnormality, the p53 protein will then emit downstream signals that either trigger the stop of cell proliferation or cell death.
Cancer cells experience many of these physiologic stresses during their development and the only way they can survive is if they shed or jettison the p53 protein. Once p53 is inactivated then this entire alarm system is deactivated and as a consequence now cancer cells can proliferate unrelentingly without having to worry about interference from p53.
Could you give a similar overview of angiogenesis?
The proliferation of a cancer cell in the presence of oncogenes and inactivated tumor suppressor genes will go on for a little while, but after a while, a very small clump of cells — let’s say 0.2 millimeters — will stop proliferating simply because the cells in the tumor mass don’t have proper access to the circulation. In the absence of that access the cells in the center of the tumor mass-small as it may be-cannot get oxygen, they cannot get nutrients from the blood, and they cannot eliminate all kinds of metabolic waste including carbon dioxide.
And so these very small tumor masses stay at that very small size. They may continue to proliferate, but they die at the same rate because they’re constantly poisoning their own nests. The only way they can break out of this predicament is if they somehow acquired direct access to capillaries, to arteries and veins, which now supply them with an abundant amount of glucose and oxygen and other nutrients that will suffice for their further replication.
And that acquired access to the circulatory system is called “angiogenesis.” How do cancer cells acquire a blood supply? They send out a series of factors, diffusible proteins that seduce capillaries to grow from the normal circulation into the tumor mass thereby supplying cells in the tumor mass with their much needed nutrients and oxygen. These so-called angiogenetic factors stimulate specialized cells in the capillaries called endothelial cells to start migrating towards the needy tumor cells and ultimately to form the capillary connections that ultimately yield glucose and oxygen.
How does all of this knowledge help doctors in diagnosis and treatment?
By understanding the molecular defects inside cancer cells we have for the first time specific markers of the malignant state. In fact, most of our diagnostic modalities to date don’t depend on all these molecular discoveries of cancer’s origins. There are many ways of discovering the presence of a tumor, most of which we could have undertaken long before we knew so much about oncogenes and tumor suppressor genes.
In the future, however, diagnostic modalities will become much more sensitive with the development of very sensitive protein and DNA assays that will be able to detect smaller and smaller numbers of barren cells in an otherwise enormous human body by comparison.
At the same time, the discovery of these molecular defects inside cancer cells provides the drug companies with attractive targets for sending in highly specific targeted compounds, anti-tumor drugs, that can attack and disable or inactivate some of the proteins that are responsible for the uncontrolled growth of cancer cells.
That promise is still one that is in a large part unrealized. Over the last quarter-century we’ve built up an enormous amount of information on understanding what makes cancer cells tick but to exploit that information and create new kinds of treatments is only starting just now. It’s a promise for the future.
So, it seems that prevention might be the best medicine?
As has been known for more than a hundred years, profound reductions in mortality from disease can be much more readily achieved by preventing the disease to begin with than by treating the disease once it arises and the same is certainly true for cancer. One could reduce cancer incidents by at least 30 or 35% of people stopped smoking tomorrow. One could probably reduce cancer incidents by another 15 or 20% if they adopted a saner diet.
These profound reductions in cancer mortality vastly outweigh what cancer researchers and drug companies can do over the next decade or two; thus it’s totally obvious that prevention is a far more effective way of reducing cancer mortality than the invention of new kinds of treatments.
Where do you see the biggest hope in treatment in the future?
The biggest hope for treatment in the future is to take advantage of the commonalities between different kinds of human cancers. For example, all solid tumors require extensive angiogenesis, and angiogenesis is a very specialized process that occurs in a slightly different way in cancer tissues than it does in normal tissues; therefore the process of angiogenesis holds the hope of creating specific targeted drugs that prevent the peculiar kind of angiogenesis that tumors carry out. Moreover, if one has such drugs, one might be able to treat in one fell swoop a wide range of solid tumors rather than only being able to treat a small narrow sector of tumors that have one or another peculiar trait.
I could cite other examples as well which persuade me that the big hope for cancer research in the future is to attack molecular and biochemical defects that are shared by a wide spectrum of tumors thereby generating cures that will be applicable to large numbers of patients.
Do you feel hopeful about the cancer field?
This is a time of enormous optimism in the field of cancer research. We now have this enormous backlog of information that we’ve accumulated for the last quarter-century and now we can begin to take advantage of it together with new and modern ways of designing drugs to really create from nothing very specific and targeted drugs. This is a period of great excitement and over the next decade, there are going to be some astounding and really stunningly successful drugs for certain kinds of cancers. That’s only just now begun.
What are the big unanswered questions in the field?
There [are] still major unanswered questions in the area of cancer research. One unanswered question is how cells that form a primary tumor acquire the ability to become invasive and metastatic. What genes change inside that cell to enable that to happen?
Another big area is the whole question of the immune system. Does the immune system represent a defense against the formation of tumors, and can we mobilize the immune system to be far more effective in attacking cancer cells than it normally is? Normally, cancers arise in individuals who have a perfectly intact immune system and somehow the cancer cells must be able to escape surveillance by the immune system and how they do that remains a major mystery.